


El tumor fibroso solitario (TFS) es una tumoración mesenquimal poco frecuente identificada inicialmente en la pleura, pero que puede presentarse en prácticamente todas las regiones. En los últimos años, se han descrito algunos casos aislados originados en el tracto genital femenino. Presentamos el caso de una mujer de 35 años de edad que consultó por una tumoración vulvar, a la que se le practicó exéresis quirúrgica. En el examen anatomopatológico la tumoración medía 20mm de diámetro, presentaba características típicas de TFS y estaba constituida por una proliferación fusocelular sin atipias, con áreas alternas hiper e hipocelulares, abundante colágena y un patrón vascular hemangiopericitomatoso. En la inmunohistoquímica era intensamente positiva para CD34 y bcl-2 y negativa para S-100 y actina. El TFS presenta un comportamiento agresivo en alrededor de un 25% de los casos y debe considerarse siempre un tumor potencialmente maligno, por lo que es preciso un seguimiento clínico. Por tanto, su correcta identificación y diferenciación de otras lesiones del área genital femenina con características similares son de importancia crucial.
Solitary fibrous tumour (SFT) is a rare mesenchymal tumour initially identified in the pleura, but that can be present in virtually all regions. Isolated cases arising in the female genital tract have been described in recent years. We report the case of a woman aged 35 who presented with a vulvar tumour, which was resected. On pathological examination the tumour measured 20mm in diameter and showed typical features of SFT, consisting of spindle cell proliferation without atypia, alternating with hyper- and hypo-cellular areas, presence of abundant collagen and with a vascular hemangiopericytomatous pattern. Immunohistochemistry was strongly positive for CD34 and bcl-2 and negative for S-100 and actin. SFT shows aggressive behaviour in approximately 25% of cases and should always be considered a potentially malignant tumour, and requires a clinical follow-up. Therefore, the identification and differentiation from other lesions with similar features in the female genital area is essential.
El tumor fibroso solitario (TFS) es una entidad clínico-patológica poco frecuente. Aunque el 70% de los casos se localizan en la región torácica pleural1,2, se han descrito numerosos casos de localización extrapleural originados prácticamente en todas los órganos3–6. En los últimos años, se han descrito algunos casos aislados en el tracto genital femenino7–9. El tumor presenta unas características clinicopatológicas peculiares y un comportamiento clínico incierto, por lo que su correcto reconocimiento resulta de crucial importancia para establecer un adecuado seguimiento3.
Se presenta un caso de tumor fibroso solitario de localización vulvar y se discute el significado clínico de estos tumores.
CASO CLÍNICOMujer de 35 años de edad, sin antecedentes de interés, que acudió a la consulta refiriendo molestia y sensación de dolor en la región genital. En el examen físico ginecológico se observó un aumento de volumen bien circunscrito en la cara posterior del labio mayor. Se practicó una exéresis quirúrgica.
El examen macroscópico reveló un fragmento de tejido de 30 mm de diámetro que al corte estaba formado por un tejido grisáceo blando, en cuyo seno se identificaba una tumoración nodular blanquecina y homogénea de 20 x 18 mm, de consistencia firme. El material de biopsia fue fijado en formalina al 10% y se incluyó en parafina; se realizaron cortes histológicos y tinción con hematoxilina y eosina para examen microscópico. Se realizó un estudio inmunohistoquímico con los anticuerpos monoclonales CD34, actina, bcl-2 y S-100 (Dako, Glostrup Dinamarca).
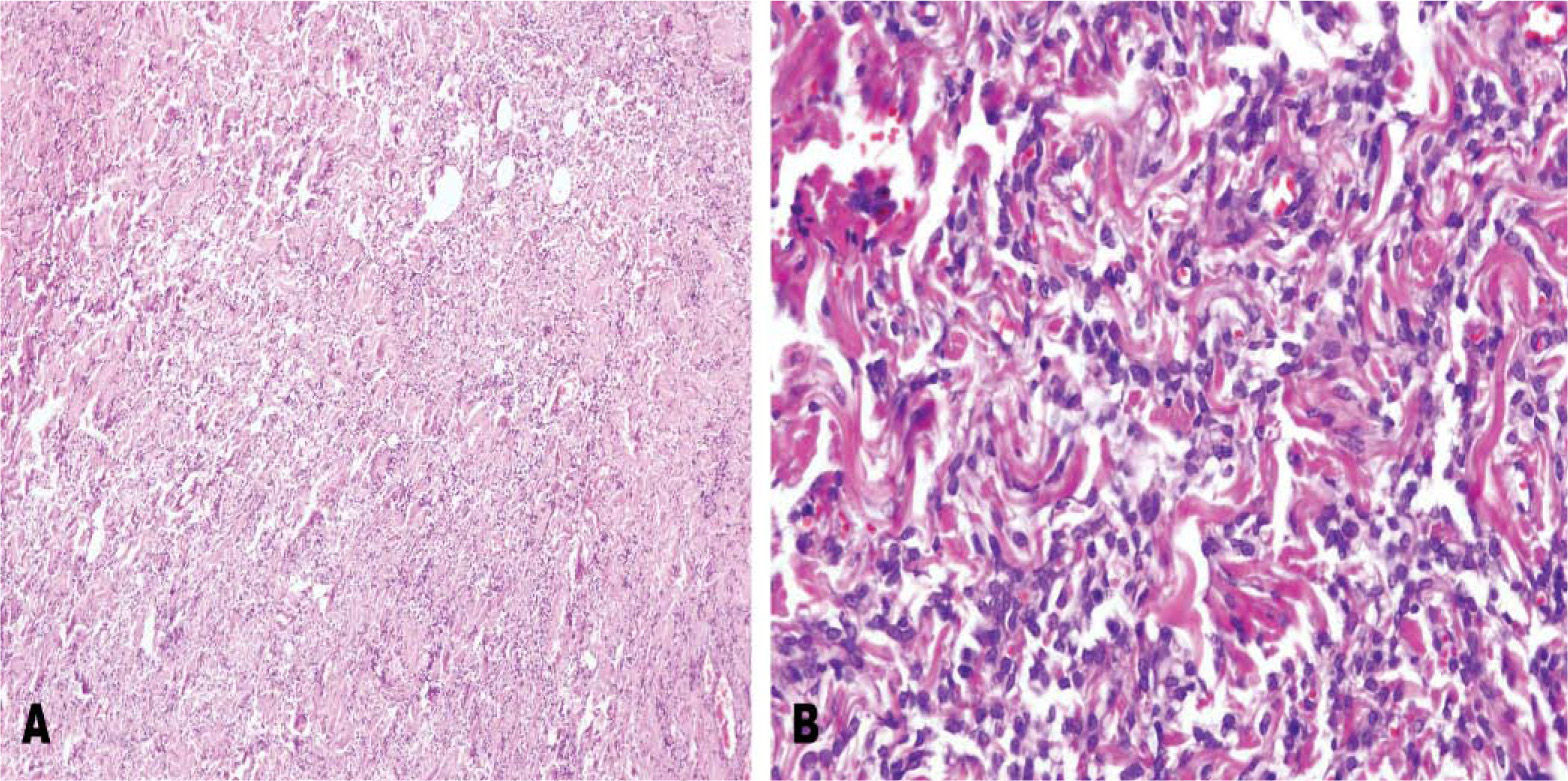
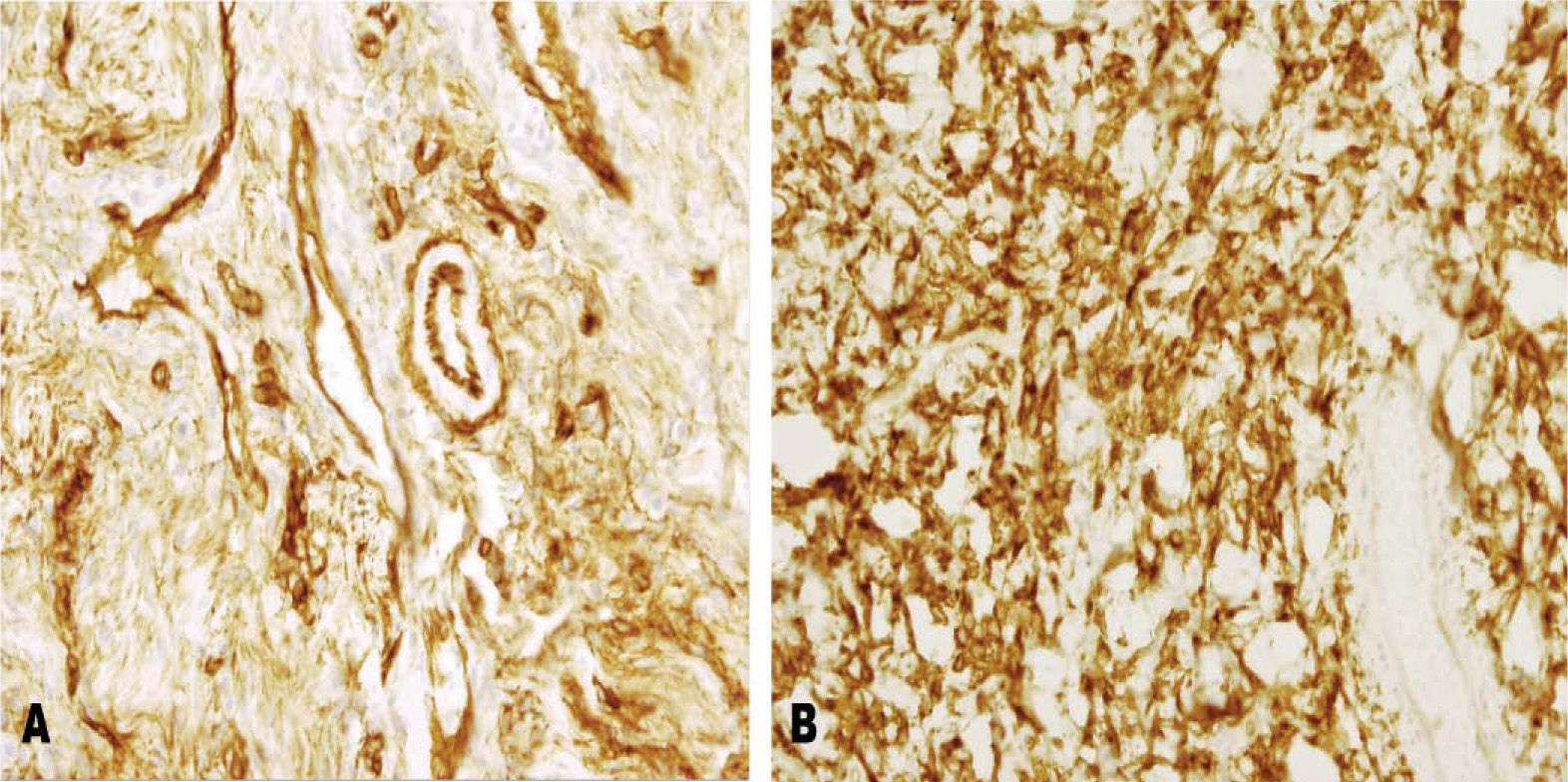
En el examen microscópico se observó una lesión predominantemente fibrosa, de borde definidos, con áreas alternas hiper e hipocelulares. Destacaba una notable proliferación de vasos de pequeño y mediano tamaño, ramificados, de paredes engrosadas y hialinizadas que conformaban un patrón hemangiopericitomatoso (fig. 1). Las células eran monomorfas, sin atipia citológica, de aspecto fusifor me y mostraban una baja actividad mitótica. Se evidenciaba abundante colágena intercelular. Los estudios inmunohistoquímicos revelaron que las células tumorales eran intensamente positivas para el antígeno CD34 y para bcl-2 (fig. 2), mientras que eran negativas para S-100 y actina.
 Proliferación de células fusiformes en citoplasma discretamente eosinófilo, proliferación vascular. B) Proliferación vascular que adoptando patrón hemangiopericitomatoide.")
 Positividad evidente de las células fusiformes a la tinción para CD34. Se observa también positividad vascular. B) Positividad intensa para la tinción con bcl-2 en las células tumorales.")
El TFS se describió por primera vez en 1931 en la pleura por Klemperer y Rabin13,10. Se consideró en principio que su génesis era a partir de las células mesoteliales y se le dio el nombre de mesotelioma fibroso benigno o solitario. El advenimiento de las técnicas inmunohistoquímicas puso de manifiesto que su origen no era en células mesoteliales, sino en células esenquimales2,6.
Aunque su localización es de forma predominante intratorácica, y especialmente en la pleura, su localización extrapleural representa el 30% de los casos1,5. Entre las localizaciones extrapleurales se han reportado casos en prácticamente todas las localizaciones, incluidos en mama11, piel4, nariz12, pulmón13, hígado14, meninge15, retroperitoneo16, ligamento redondo17, etc.5. En los últimos 10 años se han descrito varios casos en el tracto genital femenino: un caso en el cérvix uterino18, un caso en el cuerpo uterino19, un caso en la región paraovárica20, 4 casos en la vulva6,21 y 2 casos en la vagina9. Nuestro caso representa, por tanto, el décimo caso publicado de TFS de origen ginecológico.
Desde el punto de vista clínico, el tumor fibroso solitario es un tumor de crecimiento lento descubierto por regla general, como en nuestro caso, de modo incidental5. Su edad media de presentación varía entre los 5 y los 80 años, con un pico máximo durante la sexta y séptima década de vida3,4,22. Algunos tumores se han relacionado con síndromes paraneoplásicos, como hiperglucemia, o hipercalcemia19,23,24.
Desde el punto de vista anatomopatológico el TFS presenta, en general, una buena delimitación. Está constituido por una proliferación de células mesenquimatosas fusiformes separadas por bandas de colágena gruesas, y muestra a menudo áreas hialinas extensas y degeneración mixoide. Con gran frecuencia presenta zonas hipo e hipercelulares y es característica la presencia de abundantes vasos con frecuente esclerosis perivascular y un patrón vascular hemangiopericitomatoide. La atipia citológica es característicamente leve. Debido a su poca frecuencia, a la variedad de patrones histológicos y a que comparte características histológicas similares con otras lesiones, plantea frecuentemente dificultades diagnósticas7–9,17,19,21,25. Sin embargo, el TFS presenta una diferenciación inmunofenotípica característica que ayuda a establecer su diagnóstico y diferenciarlo de otras lesiones. Así, es característica de este tumor la positividad para CD34 y para bcl-2, así como la negatividad para marcadores de músculo liso (desmina, actina) y de nervio periférico (S-100). Este patrón inmunofenotípico permite confirmar el diagnóstico2,6,22.
Aunque inicialmente se clasificó el TFS como un tumor benigno, la experiencia posterior demostró que aproximadamente un 23 % de los casos presentaba un comportamiento maligno pudiendo ocasionar la muerte del paciente por recidivas locales o metástasis a distancia26. En la actualidad, se considera que el comportamiento biológico del tumor es incierto y siempre potencialmente maligno. Por tanto, un correcto diagnóstico de esta lesión es de importancia crucial, ya que se debe indicar un seguimiento estricto de las pacientes12,20,27. Las características morfológicas diagnósticas no permiten determinar el riesgo de progresión de este28.