


Los tumores neuroendocrinos de la mama constituyen una entidad poco frecuente. La existencia de marcadores neuroendocrinos ha permitido su diferenciación del resto de tumores de la mama desde hace tan solo 10 años. La mayoría son tumores de buen pronóstico que expresan receptores hormonales. Una vez descartado el origen metastásico del tumor, el tratamiento no difiere del tratamiento de otro tipo de cáncer mamario, aplicando el tratamiento adyuvante en función de los factores pronósticos y el estadio tumoral al diagnóstico. Presentamos el caso de un tumor neuroendocrino de la mama como hallazgo postoperatorio tras un tratamiento conservador de la mama por un carcinoma ductal infiltrante.
Neuroendocrine tumors of the breast are uncommon. For the last 10 years, the existence of neuroendocrine markers has allowed these tumors to be differentiated from other tumors of the breast. Most are tumors with a good prognosis that express hormone receptors. Once a metastatic origin has been excluded, treatment does not differ from that of other types of breast cancer, consisting of adjuvant therapy based on prognostic factors and tumoral stage at diagnosis. We report a case of neuroendocrine tumor of the breast as a postoperative finding after conservative treatment for infiltrating ductal carcinoma of the breast.
Los tumores neuroendocrinos o carcinoides representan menos del 1% de la totalidad de los tumores malignos de la glándula mamaria, de los cuales el 36% son lesiones metastásicas de origen extramamario. Desde el punto de vista biológico, estos tumores tienden a ser menos agresivos que el carcinoma ductal infiltrante convencional, siendo rara la presencia de enfermedad metastásica axilar. Los tumores neuroendocrinos de la mama no presentan una determinada característica clínica o radiológica y, por lo general, tampoco van acompañados de manifestaciones clínicas sistémicas de secreción hormonal relacionadas con la secreción de hormonas vasoactivas, siendo necesario para su diagnóstico que más del 50% del tumor presente marcadores neuroendocrinos.
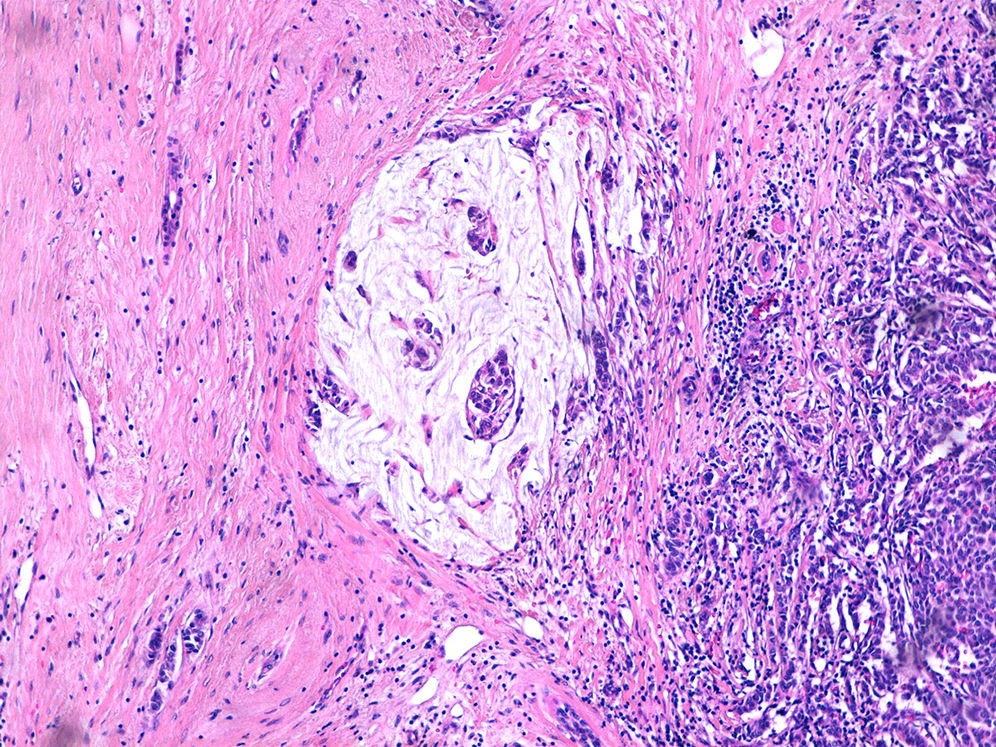
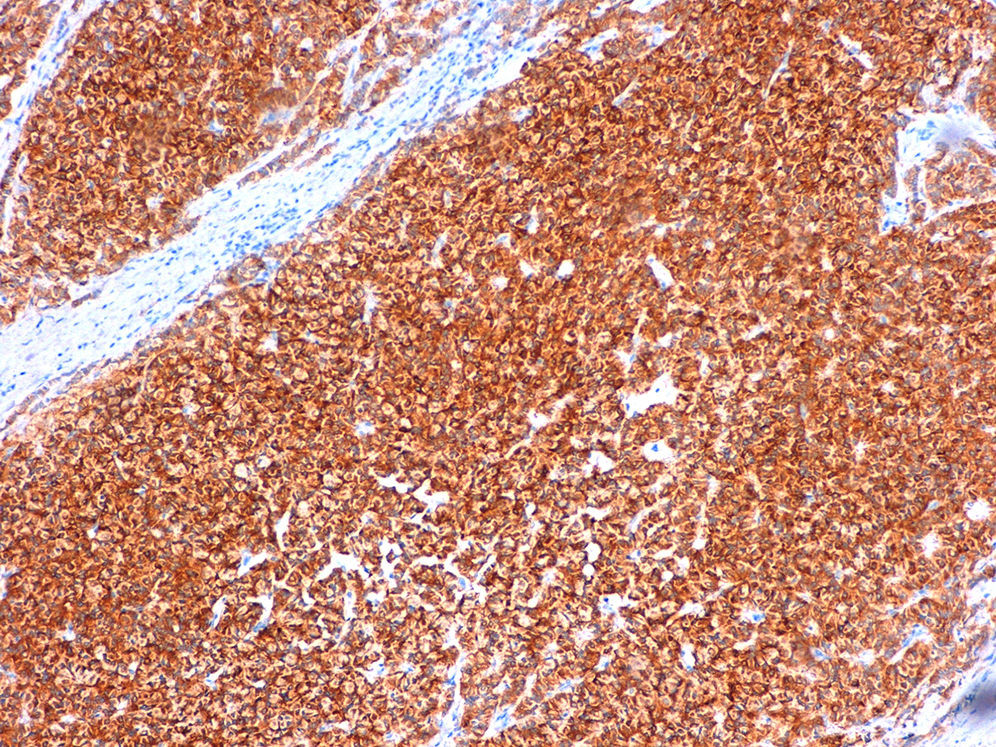
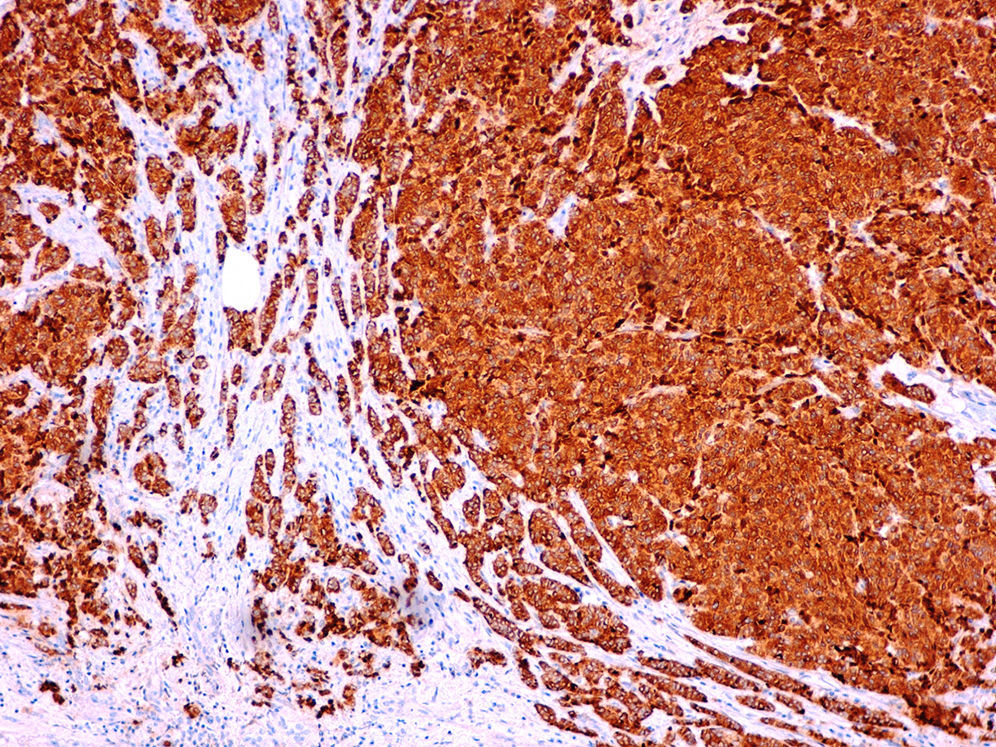
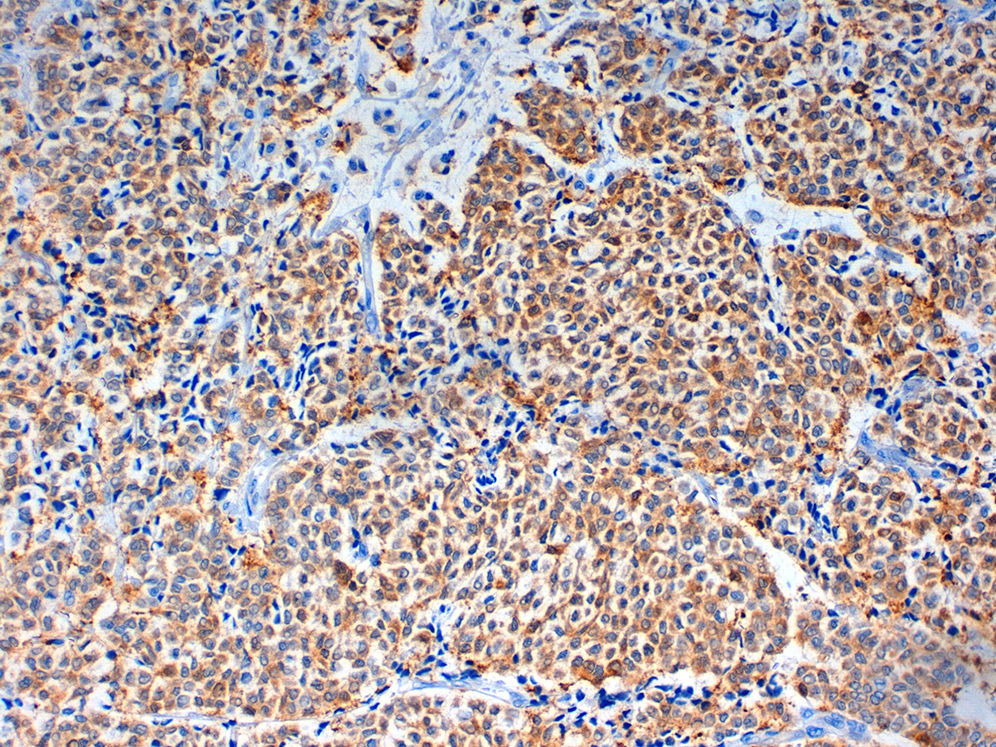
Caso clínicoMujer de 60 años, diabética insulinodependiente, hipertensa, con hipotiroidismo, dislipidemia y periartritis escápulo-humeral. Como antecedentes quirúrgicos presenta una histerectomía total abdominal a los 48 años por útero miomatoso, desde la cual ha estado en tratamiento hormonal sustitutivo (durante 7 años), laminectomía L4-L5, microcirugía laríngea por nódulos, cerclaje cervical y legrado. Ha tenido 2 embarazos, el primero de ellos a los 25 años, y no ha dado lactancia. La paciente es remitida a nuestra consulta de cirugía ginecológica desde el programa de detección precoz del cáncer de mama tras detectarse en la última mamografía un crecimiento de un nódulo, anteriormente estable, en cuadrante súpero-interno de mama izquierda, lesión catalogada como BIRADS 3. La ecografía habla de un nódulo sospechoso BIRADS 4, mientras que la resonancia magnética nuclear muestra un nódulo de 1,88×1,78×1,89cm (BIRADS 5) junto con 3 adenopatías axilares sospechosas de metástasis que no se biopsian por proximidad a vasos axilares (T1c, N1, M0). A la exploración, se palpa un nódulo de 2×2cm en cuadrante superointerno de mama izquierda, móvil y las zonas ganglionares son negativas. La biopsia del nódulo muestra un carcinoma ductal infiltrante (CDI) con receptores de estrógenos positivos al 70%, de progesterona menor al 5%, Ki 67 del 2%, p53 negativo y HER 2 neu dudoso (2+). Tras estos resultados, se programa a la paciente para intervención quirúrgica, realizándose una segmentectomía mediante mamoplastia horizontal más biopsia selectiva de ganglio centinela (BSGC) que resulta negativa. El resultado anatomopatológico definitivo viene informado como carcinoma neuroendocrino invasivo grado 2 bien diferenciado de 20mm con componente mucinoso focal (fig. 1) y cambios de tipo sólido-papilar, bordes quirúrgicos libres e invasión linfovascular presente. Se estadifica como pT1c, pNo, Mx (estadio 1A). La inmunohistoquímica presenta positividad para los receptores hormonales y negatividad para el Ki67, p53 y HER 2 Neu (comprobado mediante FISH). La sinaptofisina (fig. 2), cromogranina A (fig. 3) y enolasa neuroespecífica (fig. 4), así como CK7 y e-cadherina son positivos en más de un 50%. Como tratamiento adyuvante, la paciente recibe quimioterapia mediante 4 ciclos de epirrubicina, fluorouracilo y ciclofosfamida, seguida de radioterapia y hormonoterapia con anastrozol durante 5 años.
Discusión



El primer tumor carcinoide de la mama fue descrito en 1963 por Feyrer et al. cuando descubrieron que varios tumores invasivos de la mama presentaban ciertos patrones de crecimiento carcinoide1. En 1982 Azzopardi et al. acuñaron el término de tumores argirófilos de la mama para referirse a aquellos tumores que presentaban gránulos neurosecretores2; sin embargo, no fue hasta finales de los años 80 cuando se descubrió que estos tumores además expresaban marcadores de cromogranina y sinaptofisina3. Fueron Sapino et al. quienes, en 2002, sugirieron la primera definición de tumor neuroendocrino de la mama a partir de la expresión de una serie de marcadores4, y posteriormente la Organización Mundial de la Salud (OMS) en 2003 reconoció a los tumores neuroendocrinos de la mama como un tipo diferenciado de cáncer de mama. La OMS definió los tumores neuroendocrinos de la mama como aquellos en los que uno o varios marcadores neuroendocrinos se expresan en al menos el 50% de las células: cromogranina A o B y sinaptofisina (las más específicas) y otros como la enolasa neuroespecífica, CD56, triple proteína neurofilamento, bombín y Leu5. Cuando la expresión de estos marcadores neuroendocrinos es menor del 50%, se habla de diferenciación focal neuroendocrina, y se ha de considerar como otro tipo diferente de tumor. Para considerar el tumor como primario de la mama, es necesario haber descartado la presencia de otros tumores primarios en cualquier localización del cuerpo, así como haber encontrado un componente in situ en el tumor6.
La incidencia de estos tumores es realmente baja, entre un 0,3 y un 0,5%7, y hasta el año 2012 tan solo existía un total de 83 casos recogidos en la literatura. El 60,2% de estos tumores se dan en pacientes ≥ 50 años8.
El origen de estos tumores es aún controvertido. Inicialmente, existían 2 teorías: algunos consideraban que las células argirófilas procedían de la cresta neural y migraban hasta los ductos mamarios, mientras que otros eran partidarios de la existencia de células neuroendocrinas en la mama, a partir de las cuales se desarrollaba el componente in situ9. La hipótesis más novedosa indica una diferenciación de las células madre durante el proceso de carcinogénesis en 2 líneas: epitelial y endocrina (no se ha demostrado que los tumores de origen lobular tengan diferenciación neuroendocrina). Esta hipótesis está respaldada por los estudios moleculares que descubren que el componente neuroendocrino es idéntico al componente intraductal y todo el tumor pertenece a la estirpe luminal10. Nuestro caso aporta el caso de un tumor luminal A, a pesar de que la expresión de receptores de progesterona era tan solo del 5%, su expresión se considera positiva por encima del 1%.
La OMS reconoce 3 tipos de tumores neuroendocrinos de la mama: sólido (más frecuente), de célula pequeña y de célula grande. Algunos autores también consideran el tipo mucinoso y, aunque la OMS no ha incluido esta variante entre sus tipos histológicos, sí que reconoce que hasta el 26% de estos tumores presentan diferenciación mucinosa5. La mayoría de los tumores neuroendocrinos presentan receptores de estrógeno y progesterona positivos y Her 2 neu negativo (Luminal A)10; sin embargo, se han llegado a describir algunos subtipos basal-like (triples negativos)11.
El componente in situ del tumor neuroendocrino de la mama es importante a la hora de determinar el tratamiento. El carcinoma ductal in situ (CDIS) suele ser infradiagnosticado preoperatoriamente, y en muchos casos se asemeja a hiperplasias ductales y papilomas intraductales, siendo necesaria su exéresis quirúrgica para el diagnóstico definitivo en más de 2 tercios de los casos12.
El pronóstico no depende del tamaño del componente neuroendocrino, sino que, al igual que los demás tipos de tumores de la mama, este viene principalmente determinado por el grado histológico, así como por la producción de mucina y la diferenciación apocrina. Se consideran tumores de buen pronóstico por su buena diferenciación los de tipo sólido, los mucinosos, y los de diferenciación apocrina, mientras que los de célula grande y pequeña son tumores más indiferenciados y, por tanto, asociados a un peor pronóstico y menor supervivencia8,13. En nuestro caso se trataba de un tumor de 2cm de buen pronóstico, ya que su mayoría correspondía a tipo sólido-papilar y presentaba un componente mucinoso focal.
Existe poca información en la literatura acerca del tratamiento óptimo de estos tumores, pero es importante mencionar que el diagnóstico histológico de un tumor carcinoide de la mama no contraindica un tratamiento preservador, siempre y cuando la paciente cumpla los criterios oncológicos para este procedimiento. Si el resultado histológico es compatible con un tumor neuroendocrino, es imprescindible evaluar si la paciente tiene el antecedente de un tumor carcinoide extramamario, de no existir el antecedente, se ha de realizar una evaluación para descartar que se trate de una lesión metastásica en la glándula mamaria, lo que evitará así realizar una mastectomía innecesaria. Una vez descartados el antecedente y la presencia de un tumor neuroendocrino extramamario, el tratamiento tanto quirúrgico como adyuvante será llevado a cabo igual que en otro tipo de tumor mamario, en función de su grado histológico y extensión, tanto local como a distancia14,15. La decisión de indicar tratamientos adyuvantes locales o sistémicos se harán tomando en consideración los factores pronósticos y el estadio de la enfermedad al momento del diagnóstico, similar a como se procede en los otros tipos histológicos de carcinoma mamario. La combinación de fármacos más adecuada aún no ha sido determinada ya que, a día de hoy, aún no se han llevado a cabo ensayos clínicos controlados y aleatorizados comparando los diferentes regímenes de quimioterapia y sus resultados en este tipo de tumores8. En nuestro caso, a pesar de tratarse de un tumor de buen pronóstico, fue necesario administrar quimioterapia adyuvante más radioterapia pues el tamaño del tumor llegaba a los 2cm y se realizó tratamiento conservador.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecimiento al servicio de Anatomía Patológica del Hospital Torrecárdenas por haber cedido las imágenes que complementan el caso.