





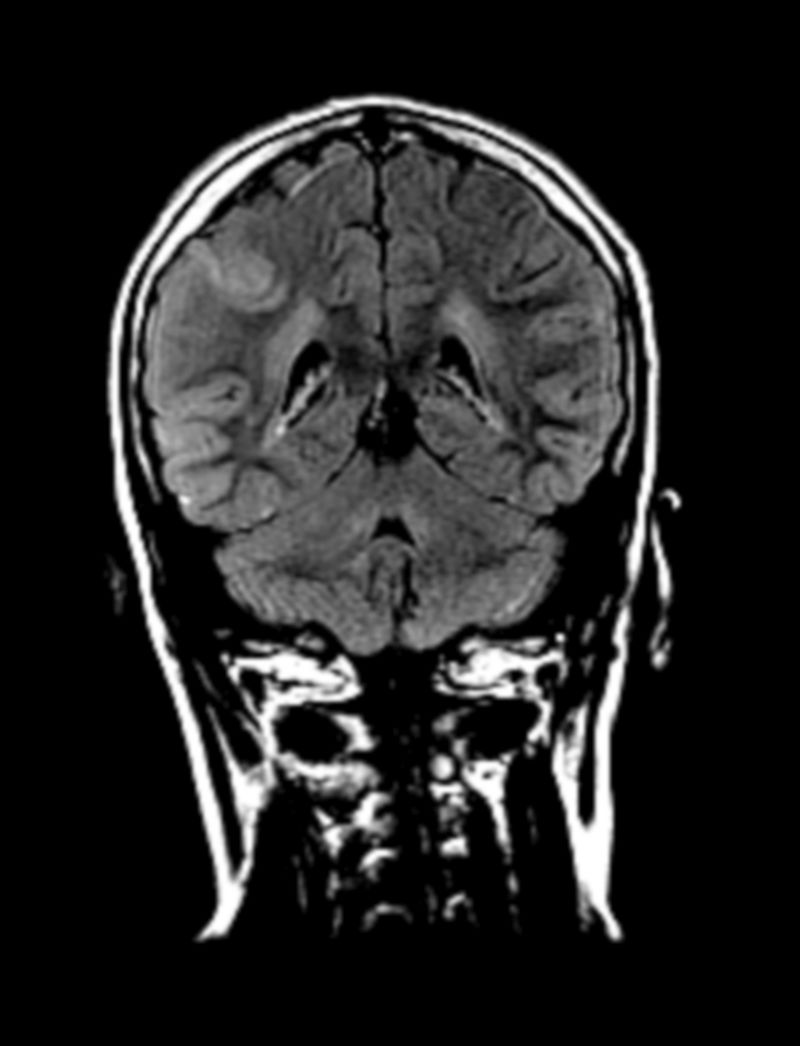
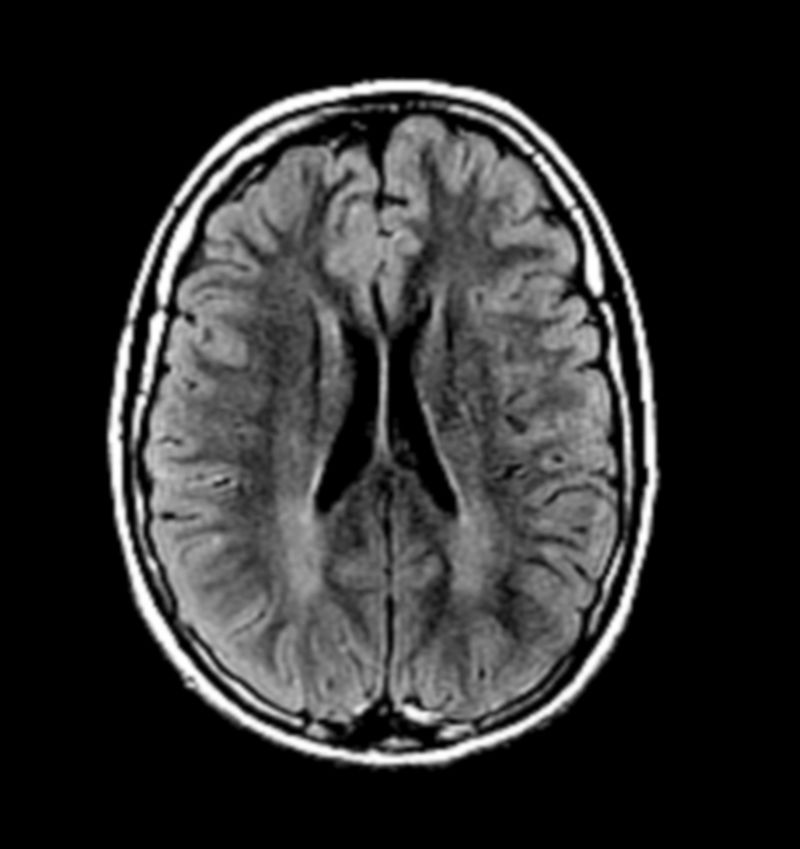
Se presenta el caso de un paciente con fenotipo conductual cambiante y biotipo llamativo, con diagnóstico de trastorno por déficit de atención e hiperactividad (TDAH) desde la infancia y con respuesta parcial a los tratamientos pautados (metilfenidato de liberación prolongada, risperidona, sertralina). Tras un episodio de agresividad en el domicilio, ingresa en una unidad de hospitalización infantojuvenil y se objetivan alteraciones en la resonancia magnética nuclear (RMN) cerebral y en el estudio metabólico, siendo estas últimas compatibles con un déficit de ornitina transcarbamilasa (OTC), uno de los enzimas del ciclo de la urea. Se discuten los distintos diagnósticos planteados en vista del cuadro sindrómico del paciente, tanto a nivel orgánico como de una comorbilidad psiquiátrica. El déficit de OTC tiene una incidencia baja, de uno por cada 15.000 recién nacidos, similar a la de la fenilcetonuria (para la cual hay un programa establecido desde hace años de cribado neonatal). Se trata de una enfermedad predominantemente hereditaria (transmisión ligada al cromosoma X) pero con numerosos casos descritos de deleciones o mutaciones de novo. Un déficit completo inicia síntomas en los primeros años de vida, mientras que un déficit parcial podría comenzar en la infancia tardía o incluso en la adolescencia, con síntomas neuropsiquiátricos o conductuales inespecíficos, no existiendo descripción de un patrón claro.
The case is presented of a patient with a changing behavioural phenotype and a rare biotype, who was diagnosed with attention deficit hyperactivity disorder (ADHD) from an early age, with partial response to the prescribed treatment (extended-release methylphenidate, risperidone, sertraline). After an aggressive episode at home, he was admitted to the child and adolescent hospitalisation unit. A brain NMR scan and a metabolic study revealed anomalies compatible with a deficiency of ornithine transcarbamylase (OTC), an enzyme which forms part of the urea cycle. The study discusses the different diagnoses that were proposed given the syndrome of the patient, both from an organic approach, as well as and from the perspective of psychiatric comorbidity. OTC deficiency has a low incidence —one in every 15,000 newborn babies—, similar to that of phenylketonuria (which was included years ago in the newborn screening program in Spain). This is a predominantly hereditary disease (X-linked transmission), but with several described cases of de novo mutations or deletions. Complete OTC deficiency shows its symptoms in the first years of life, whereas a partial deficiency could start during late childhood or even in adolescence, with unspecific neuropsychiatric or behavioural symptoms. A clear pattern has not been established.
Artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora