





Las enfermedades neurodegenerativas que cursan con demencia constituyen un problema de salud pública cada vez más importante, especialmente por el envejecimiento de la población. La radiología aporta datos diagnósticos y pronósticos significativos en estos procesos que es necesario conocer. Recientemente se han modificado algunas de las clasificaciones de estas enfermedades, incorporando sobre todo criterios patológicos, inmunohistoquímicos y genéticos. En este trabajo se revisan los distintos aspectos radiológicos y patológicos de las principales enfermedades que cursan con demencia. Para ello se aporta nuestra experiencia con los cerebros del banco de tejidos neurológicos de nuestra comunidad, a los que se les realizaron estudios de resonancia magnética posmortem antes del estudio histológico. Las imágenes de correlación radiopatológica de estos estudios se presentan para ilustrar la exposición.
Neurodegenerative diseases that course with dementia represent a public health problem that is growing in importance, mostly due to the aging of the population. Imaging techniques provide essential diagnostic and prognostic information. Some classifications of these diseases have recently been modified through the incorporation of histological, immunohistochemical, and genetic criteria. This article reviews the different radiological and pathological aspects of the most common diseases that course with dementia. We studied brains from our community's tissue bank with postmortem MRI and posterior histological examination and illustrate this exposition with images from the radiopathologic correlation.
Con la edad se producen una serie de cambios en el parénquima cerebral que se conocen como envejecimiento cerebral. La valoración de estos cambios como fisiológicos o patológicos es subjetiva y objeto de controversia. En la resonancia magnética (RM) los hallazgos relacionados con la edad se pueden dividir en 3 apartados: la atrofia cerebral, las alteraciones de la sustancia blanca y el depósito de hierro1.
La atrofia cerebral se define como una disminución del parénquima cerebral y un aumento relativo del espacio extraxial que se manifiesta como un incremento en la profundidad de los surcos y del tamaño del sistema ventricular. Esta pérdida de volumen se produce a expensas de una pérdida neuronal, pero también de una disminución de la sustancia blanca, como confirman recientes estudios de difusión por RM2.
Las alteraciones de señal en la sustancia blanca, que se manifiestan como imágenes con prolongación de señal en las secuencias T2, se conocen con distintos nombres, como leucoaraiosis, hiperintensidades de sustancia blanca, leucopatía vascular, etc. En la base de estas lesiones se encuentran una serie de alteraciones patológicas, como la pérdida de mielina o espongiosis, el aumento de los espacios perivasculares y la hialinosis de la pared de los vasos de pequeño calibre. Acerca de estos hallazgos se profundizará en el apartado de la demencia vascular. En la evaluación radiológica de estas alteraciones se puede valorar el número y tamaño de las lesiones, así como su distribución, especialmente dividida entre la región periventricular y la sustancia blanca subcortical. Se han descrito diversas escalas, como la de Fazekas o la de Sheltens3,4, especialmente utilizadas en estudios epidemiológicos, aunque en la práctica clínica la valoración más habitual son las diferencia entre alteración leve, moderada y grave, en función del número y carácter confluente de las lesiones1,5.
El tercer hallazgo relacionado con el envejecimiento cerebral es el depósito de hierro, que se identifica como zonas de acortamiento de señal en las secuencias T2 y se hace más patente en las secuencias eco de gradiente por su mayor sensibilidad a los artefactos de susceptibilidad magnética. El hierro se empieza a depositar a partir de los 25 años en el globo pálido y, posteriormente, en el núcleo rojo, sustancia negra, núcleos dentados, caudado y putamen. También se deposita en la corteza cerebral, especialmente en el córtex motor1.
Asimismo, los hallazgos descritos anteriormente se relacionan con factores de riesgo vascular (hipertensión arterial [HTA], tabaco, dislipemias, etc.) y se asocian con enfermedades neurodegenerativas. Con la edad aumenta la prevalencia tanto de los factores de riesgo vascular como de los procesos degenerativos, lo que complica la interpretación de estas alteraciones.
Demencia: cuadro clínico y clasificaciónLa demencia es un síndrome clínico de deterioro cognitivo adquirido que se produce por una disfunción cerebral. Según los criterios del Diagnostic and Statistical Manual (DSM IV)6 se define como un deterioro de la memoria a corto y largo plazo, asociado a un deterioro del pensamiento abstracto, deterioro del juicio, a otros trastornos de la función cortical superior o a un cambio de personalidad. El trastorno es lo suficientemente grave como para interferir significativamente en el trabajo o actividades sociales habituales del paciente o en sus relaciones con los demás.
Es decir, para su diagnóstico es importante la presencia de las siguientes características clínicas: inicio gradual, declive progresivo, pérdida de memoria, deterioro de otros dominios cognitivos e interferencia con la función habitual7. Un concepto clínico importante es el de deterioro cognitivo leve (DCL), en el que se produce un déficit de memoria con otros dominios cognitivos y actividades de la vida diaria intactos.
Si el cuadro de deterioro cognitivo se asocia con otros síntomas neurológicos se puede orientar el diagnóstico sindrómico. Así, la presencia de un cuadro parkinsoniano orienta hacia una degeneración corticobasal o una demencia de cuerpos de Lewy. Una alteración del lenguaje sugiere la presencia de una afasia progresiva primaria. Una apraxia o mano alienígena se asocia a la degeneración corticobasal, y la presencia de mioclonías hace pensar en una enfermedad por priones.
Las enfermedades degenerativas primarias del sistema nervioso central son un complejo grupo de procesos de difícil clasificación. Hay clasificaciones clínicas, en función del cuadro de presentación, que permiten orientar inicialmente el diagnóstico del paciente. No obstante, cada vez se tiende a tener como referencia las clasificaciones patológicas con bases inmunohistoquímicas, con especial énfasis en el tipo de proteína que se acumula y que se relaciona con el daño cerebral. Así, se habla de las amiloidopatías, en las que hay depósito patológico de β-amiloide, las taupatías, en las que se acumula la proteína tau, o las α-sinucleopatías en las que la proteína patológica es la α-sinucleína. No obstante, hay una importante limitación diagnóstica dado el solapamiento de las características patológicas de estas enfermedades, como consecuencia de su heterogeneidad así como por los cambios relacionados con la edad y la concurrencia de varios de estos cuadros8.
Tradicionalmente, los estudios de imagen cerebral se han utilizado como métodos de exclusión de causas potencialmente tratables de demencia (tumores, hematomas subdurales, hidrocefalia normotensiva, etc.). Sin embargo, estas técnicas también presentan un valor importante en el diagnóstico de varios procesos degenerativos, tanto en las imágenes estructurales, especialmente con RM (datos de atrofia, lesiones vasculares, etc.), como en la imagen funcional, sobre todo la tomografía computarizada por emisión de fotón único (SPECT) y la tomografía por emisión de positrones (PET), donde se pueden observar datos de hipoperfusión o hipometabolismo focal. Cabe destacar especialmente su papel en el diagnóstico en fases iniciales así como en los controles evolutivos de estos pacientes que permiten definir factores pronósticos y realizar evaluaciones terapéuticas9,10.
Si dentro de las enfermedades neurodegenerativas se consideran únicamente las que asocian demencia, se puede establecer la siguiente clasificación, que se seguirá en la exposición posterior:
- A.
Enfermedad de Alzheimer (EA).
- B.
Demencia vascular.
- C.
Enfermedades de cuerpos de Lewy o α-sinucleopatías.
- •
Demencia de cuerpos de Lewy.
- •
Otros: Parkinson con demencia, atrofia multisistémica, Hallervorden-Spatz.
- •
- D.
Degeneración lobular frontotemporal (v. clasificación posterior).
- E.
Otros: corea de Huntington, enfermedad de Creutzfeldt-Jakob, lipofuscinosis, etc.
El diagnóstico definitivo de las enfermedades neurodegenerativas es histológico, y en muchos procesos, el diagnóstico clínico se considera únicamente como probable. La mayor parte de los estudios radiológicos se basa en series clínicas en las que se han descrito una serie de hallazgos en estas entidades. En este trabajo se presentan, para ilustrar la exposición, casos de enfermedades degenerativas confirmados mediante estudio histológico, procedentes del banco de tejidos neurológicos de nuestra comunidad. Se exponen correlaciones entre los hallazgos patológicos (macroscópicos y microscópicos) y las imágenes obtenidas mediante RM posmortem.
En los cerebros donados al banco, se realiza, por protocolo, una congelación de un hemisferio, habitualmente el derecho (secciones coronales de 1cm a −80°C) y un estudio histológico del izquierdo. Sobre este último hemisferio se realizaron los estudios de RM, después de fijarse en formaldehído al 10% (al menos 15 días) e incluirse en una solución de agar al 1,5% para mejorar la consistencia y el contraste. En la exploración de RM se incluyeron secuencias T1 sagital (TR/TE de 500/7,8, matriz 460×512), DP-T2 axial (TR/TE de 2.800/14–82, matriz 448×512) y FLAIR coronal 3D con bloque de 160 cortes de 1mm de grosor (TI/TR/TE de 2.200/6.000/353, matriz 258×256). Esta última secuencia se utilizó para el análisis y para la obtención de las imágenes que se presentan en este trabajo.
En el estudio patológico se incluyen secciones de parafina de 4μ que se tiñen con hematoxilina-eosina y una serie de anticuerpos específicos (anti-tau, anti-β-amiloide, anti-α-sinucleína, anti-PrPSc, anti-ubicuitina y anti-TDP-43) con el fin de realizar el diagnóstico de enfermedad neurodegenerativa.
Enfermedades neurodegenerativasEnfermedad de AlzheimerLa EA es la causa más frecuente de demencia (70%). Su incidencia aumenta con la edad y llega a afectar a un 50% de las personas mayores de 85 años. Se produce un deterioro progresivo que se inicia con una pérdida aislada de la memoria. Según los criterios de NINDS-ADRDA se clasifica en EA posible, probable y definitiva. Para el diagnóstico definitivo se necesitan criterios clínicos y patológicos. El DCL se considera un factor de riesgo para el desarrollo de EA7.
La mayoría de los casos prevalentes de EA, más del 90%, son esporádicos y se manifiestan después de los 65 años. Esta enfermedad presenta una base genética en aproximadamente un 10% de los casos. Las formas familiares son las que presentan un inicio más precoz. Se han descrito varias mutaciones implicadas, como la del gen de la proteína precursora de amiloide (en el cromosoma 21), gen presenilina-1 (en el cromosoma 14) o el gen presenilina-2 (en el cromosoma 1). Recientemente se ha relacionado la EA con la apolipoproteína ε. Ésta es un componente de las lipoproteínas de la que se han identificado 3 alelos normales ε2, ε3 y ε4. El alelo más prevalente en la población general es el ε3, en aproximadamente un 80%. La forma ε4 se asocia a un riesgo elevado de desarrollar EA y con un inicio a menor edad11.
Hay 2 lesiones histológicas características de EA: la placa senil neurítica y el ovillo neurofibrilar. La placa senil neurítica está formada por un núcleo denso de β-amiloide con células inflamatorias y neuritas distróficas en su periferia. Los ovillos neurofibrilares son agregados patológicos intracitoplasmáticos de proteína tau en forma hiperfosforilada, en las neuronas. Hay una disminución en la densidad de las sinapsis y pérdida neuronal con atrofia secundaria. También se observa gliosis en la sustancia gris y la sustancia blanca, así como cambios relacionados con angiopatía amiloide cerebral7,9.
Respecto a la localización de los ovillos neurofibrilares, éstos siguen un patrón ordenado; inicialmente la afectación se encuentra en el córtex entorrinal para, posteriormente, afectar al hipocampo, áreas límbicas del lóbulo temporal, áreas de asociación del neocórtex y, por último, áreas primarias sensoriales y motoras. En este orden se basa la clasificación más utilizada de la enfermedad, la de Braak y Braak, descrita en 1991, que divide el nivel de afectación en 6 estadios12.
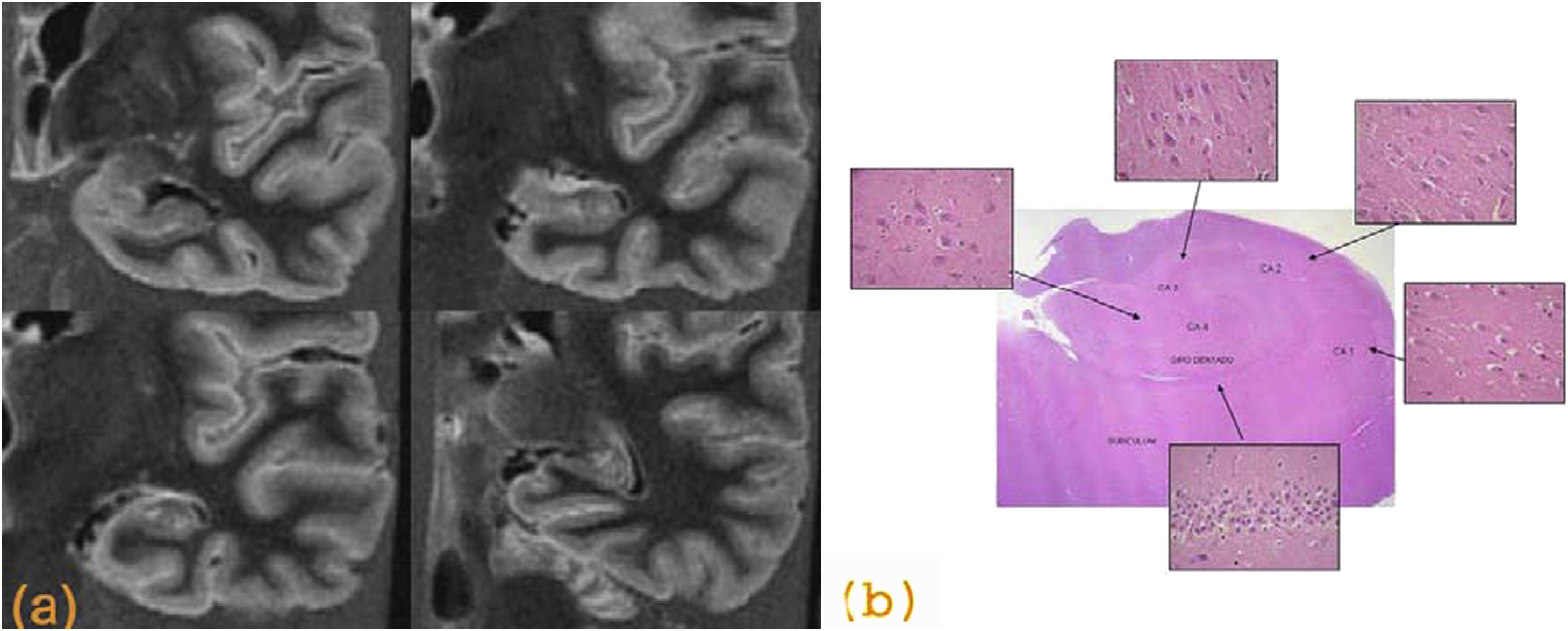
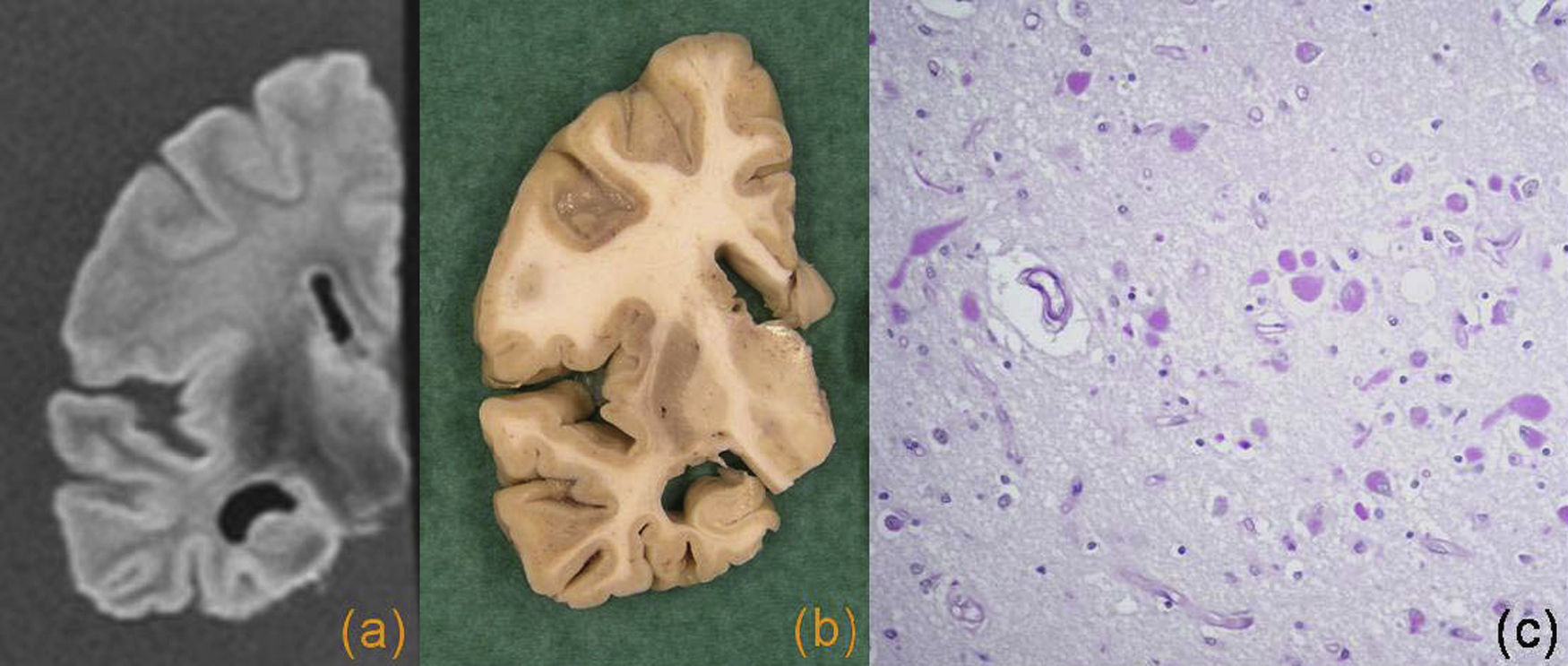
Desde el punto de vista radiológico, el hallazgo más significativo es la presencia de una atrofia, mayor en el hipocampo y el lóbulo temporal medial (figs. 1 y 2). Se puede realizar una valoración cualitativa, visual o cuantitativa, y volumétrica, esta última especialmente útil en los ensayos clínicos y en los estudios de seguimiento13. Se sabe que la medida del volumen del hipocampo permite detectar la EA en fases precoces y que se relaciona con la gravedad de la enfermedad9,14. También se ha demostrado que es un marcador de conversión a EA en pacientes con DCL15. Otros estudios han demostrado un valor similar de la medida del hipocampo y del córtex entorrinal, hallazgo significativo dado que la medición de la corteza entorrinal en las pruebas de imagen es un proceso más complejo16,17. También se ha observado una mayor incidencia de lesiones de sustancia blanca en pacientes con EA, sugiriéndose un efecto aditivo que contribuye al deterioro cognitivo18.
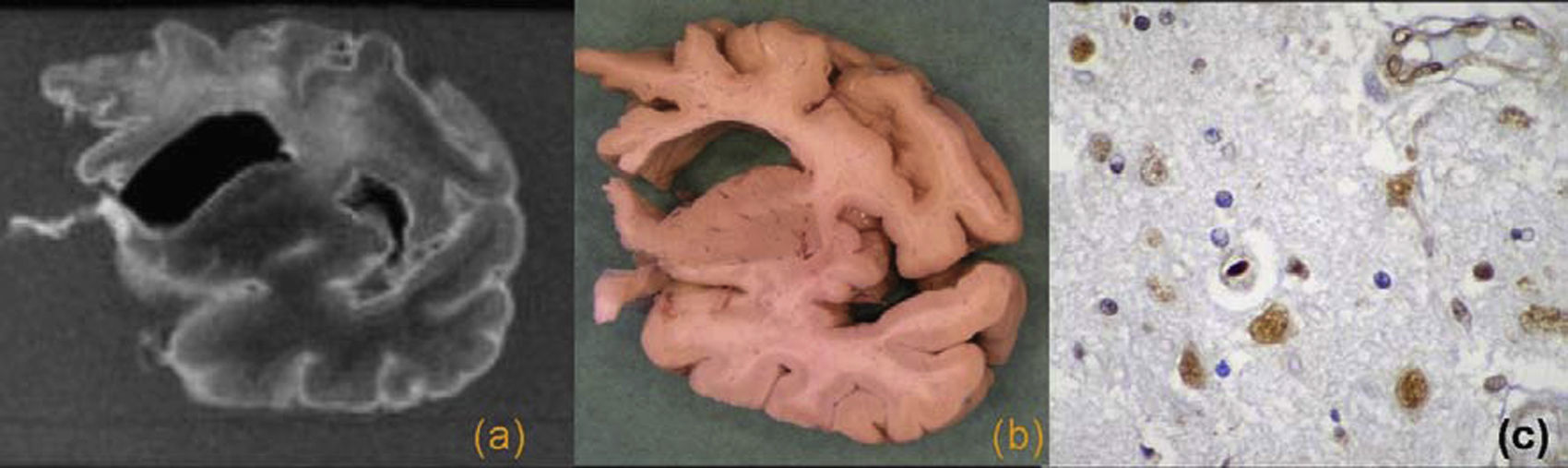
 Cortes coronales consecutivos centrados en la región temporal medial en secuencia FLAIR. b) Corte histológico del hipocampo donde se detallan las distintas áreas con su población neuronal (tinción HE).")
 y pieza macroscópica (b), donde se observa una importante atrofia del hipocampo y de la corteza entorrinal. c) Imagen microscópica del hipocampo, con tinción tau, donde se observa la presencia de múltiples ovillos neurofibrilares intraneuronales y placas neuríticas aisladas.")
Enfermedad de Alzheimer. Cortes coronales de resonancia magnética (a) y pieza macroscópica (b), donde se observa una importante atrofia del hipocampo y de la corteza entorrinal. c) Imagen microscópica del hipocampo, con tinción tau, donde se observa la presencia de múltiples ovillos neurofibrilares intraneuronales y placas neuríticas aisladas.
Las nuevas técnicas de RM se han aplicado también al estudio de la EA. Así, en espectroscopia se ha descrito una disminución significativa del N-acetilaspartato y un aumento del mioinositol19,20. En secuencias de difusión se encontró un aumento del coeficiente de difusión aparente en hipocampo y en sustancia blanca parietal21. Con estudios de perfusión se ha descrito un patrón similar al descrito en la SPECT con una disminución bilateral del flujo temporoparietal22. La PET tiene un papel muy importante en el diagnóstico de la EA y otros procesos degenerativos. Recientemente se está utilizando, sobre todo en investigación, la PET con trazadores de amiloide que puede convertirse en un biomarcador de EA23.
Demencia vascularLa demencia vascular se define como un cuadro de deterioro cognitivo secundario a enfermedad cerebrovascular. Se considera la segunda o tercera causa de demencia, aunque su diagnóstico es problemático. Desde un punto de vista clínico, se caracteriza por un inicio súbito con progresión escalonada, con presencia de datos de disfunción ejecutiva frontal. Hay datos de focalidad neurológica y criterios de imagen para su diagnóstico. Es necesario demostrar una relación temporal entre demencia y enfermedad cerebrovascular que, de forma un tanto arbitraria y discutible, se ha establecido en un período inferior a 3 meses24.
Respecto a la etiopatogenia y los hallazgos radiopatológicos se consideran dos tipos de mecanismos vasculares: la patología de vaso grande que se ha llamado clásicamente demencia multiinfarto y la patología de vaso pequeño o demencia subcortical24–26.
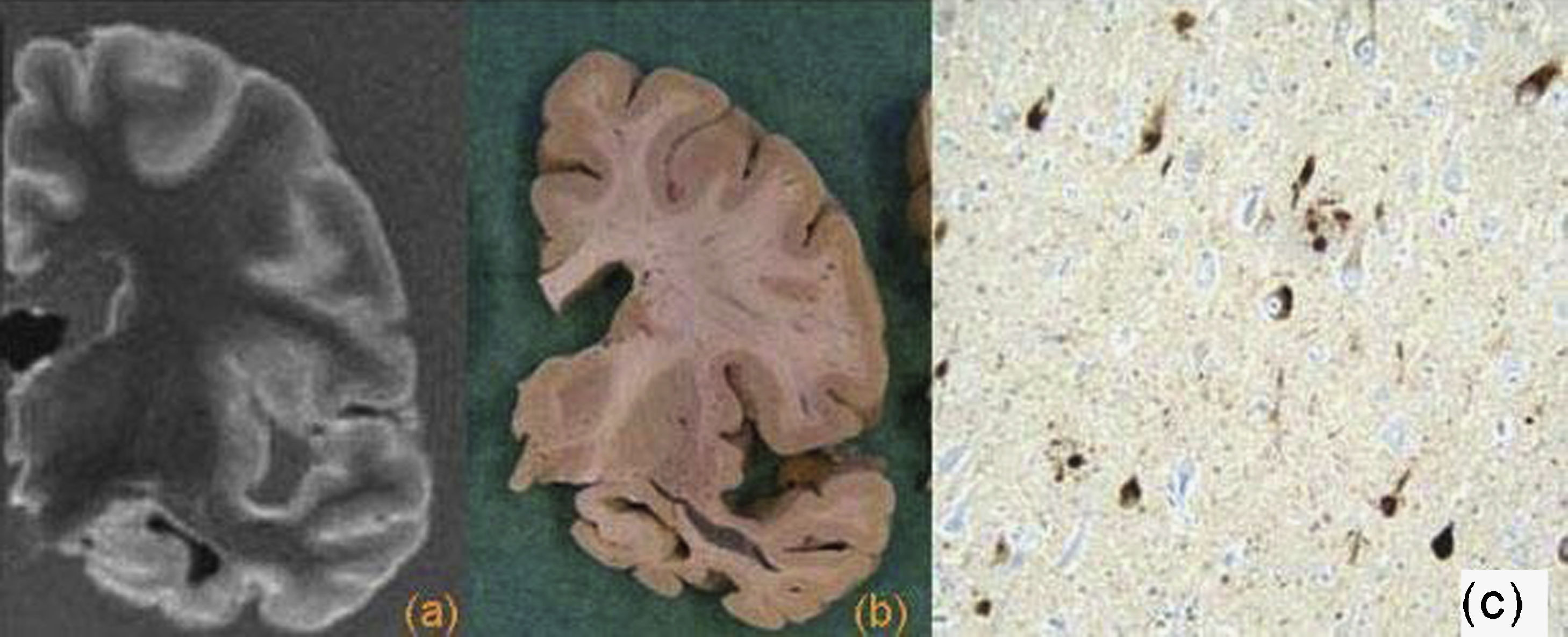
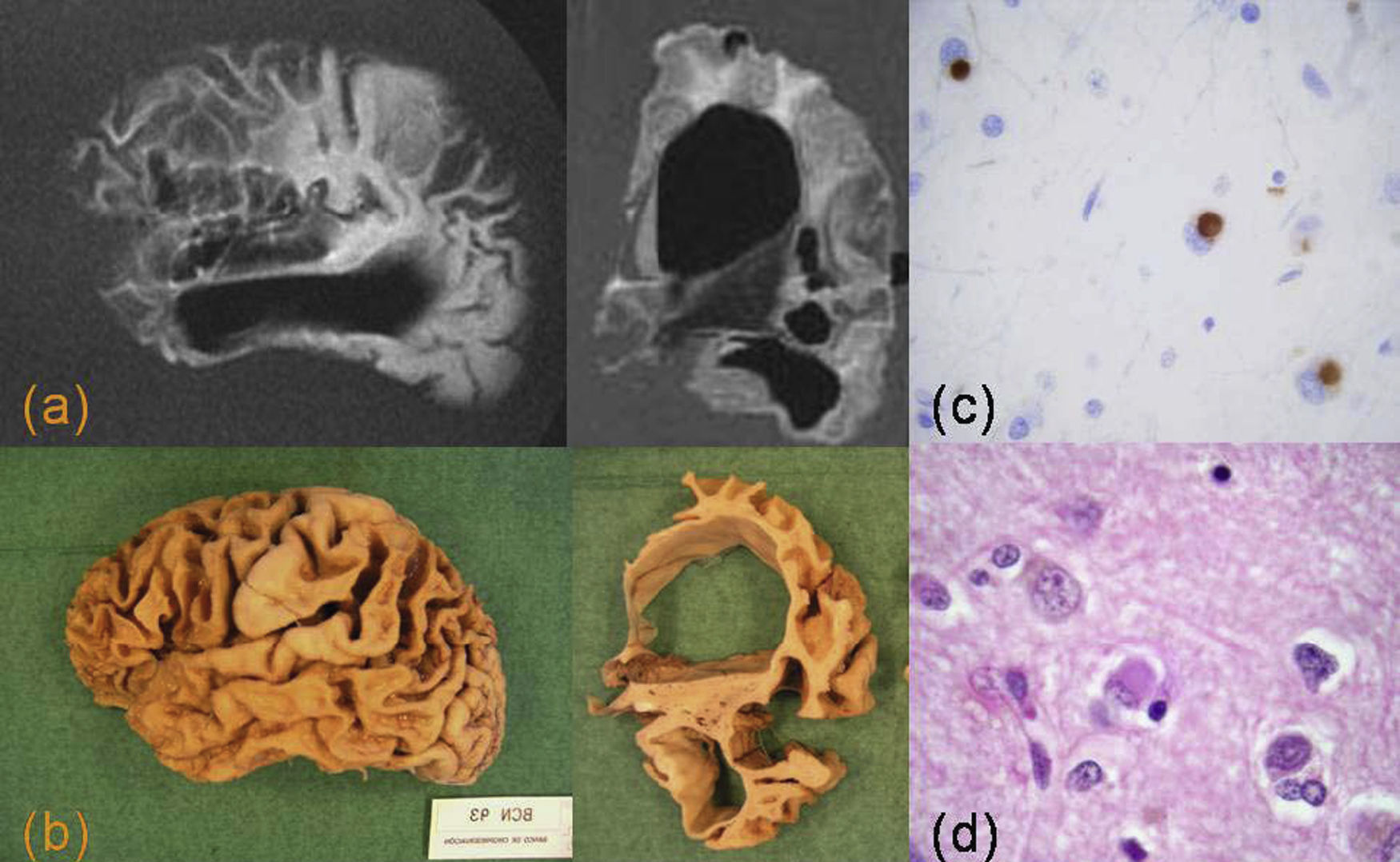
En la demencia multiinfarto se encuentran varios tipos de lesión, como infartos territoriales embólicos o ateroscleróticos, infartos frontera y necrosis cortical, secundarios a fenómenos de hipoperfusión y los microinfartos corticales a los que recientemente se les ha otorgado una mayor atención en la etiopatogenia de la enfermedad27. Estas lesiones contribuyen a la enfermedad cognitiva por 2 mecanismos: el daño acumulativo por “cantidad” de tejido dañado y la localización de éstos, los llamados infartos estratégicos como el giro angular, la corteza temporal medial bilateral, la corteza cingulada, etc. (fig. 3).
 y macroscópico (b), donde se observa un infarto crónico de la arteria cerebral anterior, con afectación de la corteza pericallosa. En la resonancia magnética se observa una hiperintensidad en la sustancias blanca y gris, y atrofia secundaria. c) En la imagen microscópica se observa una zona de necrosis, con presencia de macrófagos espumosos y astrocitos reactivos (tinción HE).")
Demencia vascular. Cortes coronales de resonancia magnética (a) y macroscópico (b), donde se observa un infarto crónico de la arteria cerebral anterior, con afectación de la corteza pericallosa. En la resonancia magnética se observa una hiperintensidad en la sustancias blanca y gris, y atrofia secundaria. c) En la imagen microscópica se observa una zona de necrosis, con presencia de macrófagos espumosos y astrocitos reactivos (tinción HE).
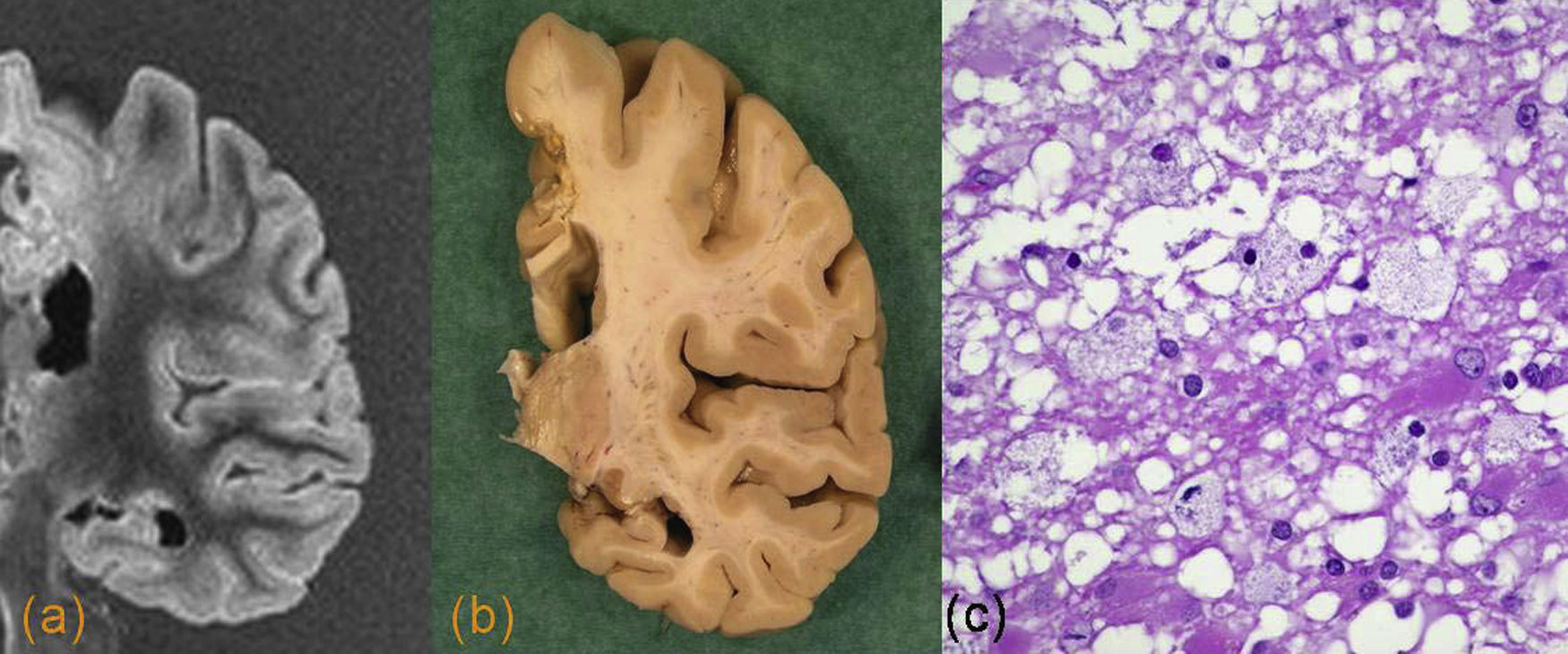
En la demencia subcortical se pueden encontrar distintos tipos de lesiones de vaso pequeño (fig. 4). Los infartos lacunares se producen por oclusión de arterias penetrantes cortas y se asocian con daño cognitivo, también cuando son numerosos o cuando presentan una localización estratégica como el tálamo (bilateral), núcleo caudado y rodilla de cápsula interna. Las microhemorragias crónicas también son marcadores de enfermedad de vaso pequeño. La isquemia de sustancia blanca se produce por afectación de arterias medulares largas que puede producir infartos y leucopatía secundaria a desmielinización vascular. Esta última entidad, también conocida como leucoaraiosis, se traduce como palidez de mielina y gliosis en histología. La isquemia crónica condiciona una pérdida de mielina y posteriormente necrosis. Esta afectación de sustancia blanca respeta la corteza y las fibras arcuatas o fibras U (dato importante en el diagnóstico diferencial radiológico con otras enfermedades de sustancia blanca, como la esclerosis múltiple, en la que sí se afectan estas fibras) y se asocia a fibrohialinosis arterial con engrosamiento de pared de los vasos (fig. 5). Se asocia a factores de riesgo como la edad y la hipertensión arterial, y también con otros, como el tabaco, la diabetes mellitus y la hipotensión postural. Su relación con el deterioro cognitivo es controvertida, aunque se acepta por la mayor parte de los autores5,25,28.
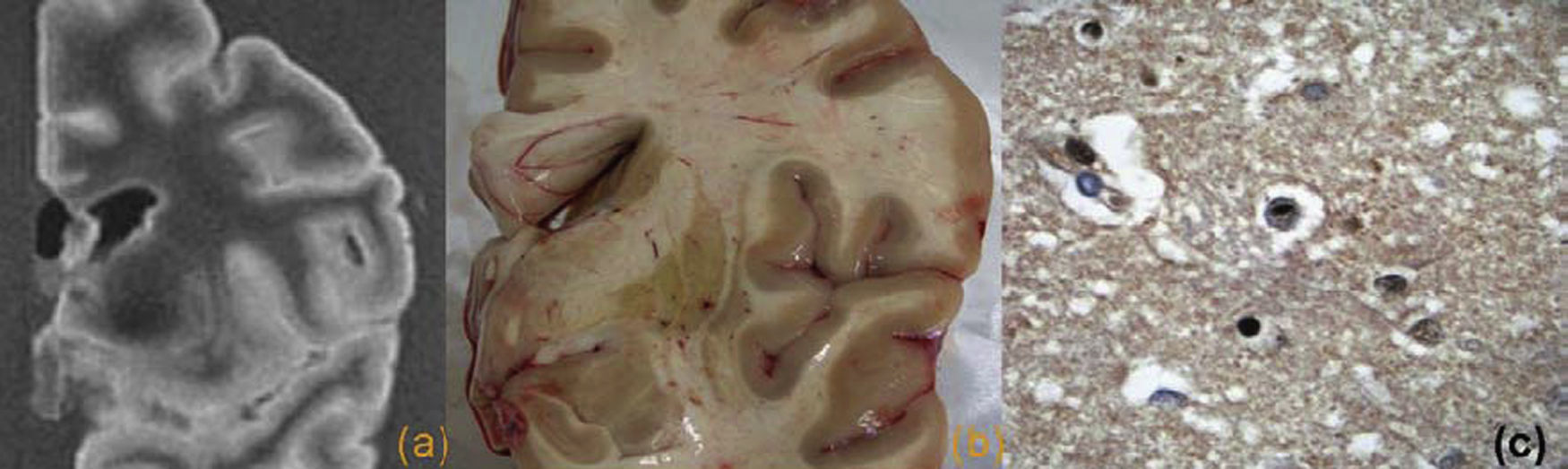
 y macroscópico (b) de un paciente con demencia y múltiples infartos lacunares en ganglios basales y sustancia blanca subcortical. c) En la imagen microscópica a bajo aumento se observa un infarto lacunar en putamen (tinción HE).")
Demencia vascular. Cortes coronales de resonancia magnética (a) y macroscópico (b) de un paciente con demencia y múltiples infartos lacunares en ganglios basales y sustancia blanca subcortical. c) En la imagen microscópica a bajo aumento se observa un infarto lacunar en putamen (tinción HE).
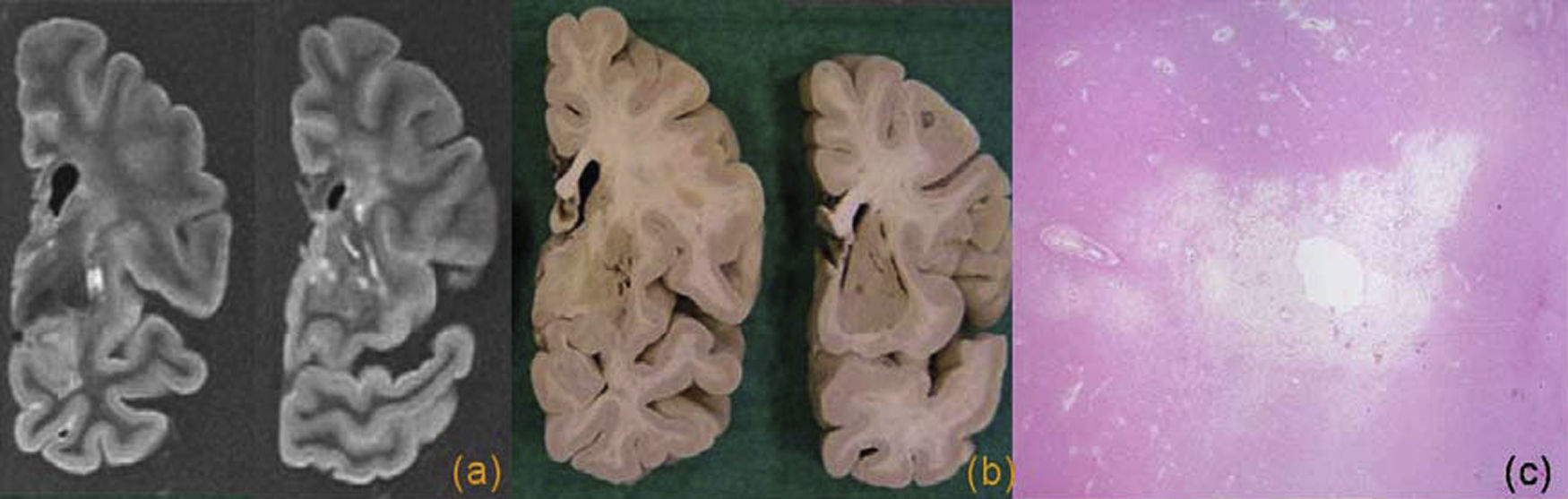
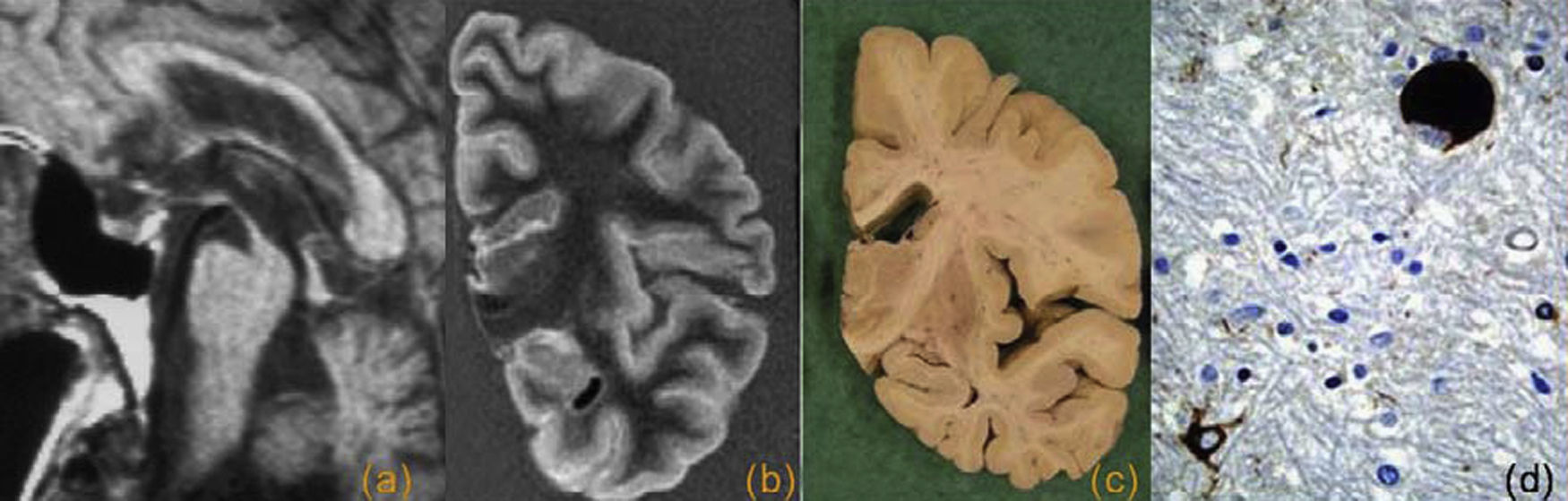
 Imagen de resonancia magnética coronal de la región frontal donde se observa una hiperintensidad de señal confluente en la sustancia blanca periventricular y subcortical. b) Imágenes microscópicas a diferentes aumentos, con tinción de mielina (tinción luxol fast blue) que muestran desmielinización. En las imágenes con tinción HE se observa hialinosis de la pared de los vasos y aumento del espacio perivascular. c) Nótese una mayor espongiosis en la región perivascular.")
Isquemia de sustancia blanca. a) Imagen de resonancia magnética coronal de la región frontal donde se observa una hiperintensidad de señal confluente en la sustancia blanca periventricular y subcortical. b) Imágenes microscópicas a diferentes aumentos, con tinción de mielina (tinción luxol fast blue) que muestran desmielinización. En las imágenes con tinción HE se observa hialinosis de la pared de los vasos y aumento del espacio perivascular. c) Nótese una mayor espongiosis en la región perivascular.
Dentro de la patología vascular asociada a demencia hay que dedicar una mención especial a la angiopatía amiloide. Ésta se caracteriza por el depósito de β-amiloide en la pared de los vasos. Se relaciona con la edad (5–23% en mayores de 60 años y hasta un 60% a los 80 años) y es más frecuente en la EA. No se relaciona con la hipertensión, aterosclerosis ni diabetes mellitus. Hay formas familiares y esporádicas. Se manifiesta como hemorragias cerebrales (es la causa más frecuente en pacientes normotensos) o como un cuadro de demencia progresiva y/o infartos pequeños múltiples26. En la anatomía patológica se encuentran depósitos eosinófilos homogéneos en las capas adventicia y media de los vasos corticales y meníngeos, lo que provoca un aumento de la fragilidad vascular. Radiológicamente se pueden ver hematomas lobulares superficiales (con extensión a la sustancia blanca o al espacio subaracnoideo), microhemorragias corticales en las secuencias eco de gradiente, y una alteración de señal de sustancia blanca.
El diagnóstico de demencia vascular es problemático y en los últimos años se ha desarrollado una controversia importante acerca de su entidad y la relación entre los mecanismos etiopatogénicos de la enfermedad vascular y las enfermedades neurodegenerativas, basados especialmente en los estudios anatomopatológicos en los que se observa una coexistencia de ambos procesos. Se sabe que hay una coadyuvancia entre EA y demencia vascular, es decir, la enfermedad degenerativa necesita menos volumen lesional para producir un deterioro clínico si se produce en un cerebro con daño vascular y viceversa, la insuficiencia vascular contribuye a la manifestación clínica de pacientes con EA preclínica. Esta interacción entre las lesiones vasculares y degenerativas hace cuestionar la existencia de la demencia vascular pura, que probablemente es menos frecuente de lo que clásicamente se le ha atribuido. Así, se ha desarrollado el término de demencia mixta que probablemente constituye una causa más frecuente de demencia29. En nuestra experiencia en el banco de tejidos neurológicos, se confirman estas tendencias, ya que el diagnóstico de demencia mixta fue el segundo en frecuencia, y el diagnóstico de demencia vascular pura fue relativamente infrecuente.
Enfermedad con cuerpos de Lewy (α-sinucleopatías)Este grupo de enfermedades se caracteriza por el acúmulo patológico de α-sinucleína. La enfermedad tipo de este grupo es la demencia de cuerpos de Lewy, que constituye aproximadamente entre un 15 y un 25% de las demencias. Clínicamente se caracterizan por la presencia de fluctuaciones cognitivas, alucinaciones visuales y parkinsonismo. Inicialmente puede no haber déficit de memoria. En el estudio histológico se encuentran cuerpos de Lewy que son inclusiones pálidas eosinófilas similares a las que se encuentran en el tronco cerebral en los pacientes con enfermedad de Parkinson. En estudios radiológicos, lo más característico es la ausencia de una atrofia marcada del lóbulo temporal medial (fig. 6). En la SPECT se presenta un patrón de hipoperfusión occipitoparietal medial y en la PET se observa un hipometabolismo dopaminérgico en el núcleo estriado30,31.
 y macroscópico (b) donde no hay atrofia significativa. Imagen microscópica donde se observa un cuerpo de Lewy cortical (tinción anti-α-sinucleína) (c) y un cuerpo de Lewy en una neurona de la sustancia negra (tinción HE) (d).")
Demencia de cuerpos de Lewy. Cortes coronales de resonancia magnética (a) y macroscópico (b) donde no hay atrofia significativa. Imagen microscópica donde se observa un cuerpo de Lewy cortical (tinción anti-α-sinucleína) (c) y un cuerpo de Lewy en una neurona de la sustancia negra (tinción HE) (d).
La enfermedad de Parkinson con demencia presenta una clínica y una neuropatología común con la demencia con cuerpos de Lewy aunque con mayor afectación de la sustancia negra. Se considera el período de 1 año para distinguirla de los pacientes con Parkinson que desarrollan demencia en el curso de la enfermedad (hasta un 40%)32.
La atrofia multisistema (AMS) es otra enfermedad de este grupo que puede asociar un cuadro de demencia. Sus características clínicas incluyen parkinsonismo, signos cerebelosos y piramidales, y fallo autonómico. Se consideran 2 subtipos: la AMS-P con síntomas parkinsonianos predominantes (anteriormente conocida como degeneración estriatonigral), que constituye el 80% de los casos, y la AMS-C, con predominio de síntomas cerebelosos (antes denominada atrofia olivopontocerebelosa), que constituye un 20%. En anatomía patológica se demuestran inclusiones con α-sinucleína en células oligodendrogliales. En radiología se ha descrito una atrofia del núcleo estriado, sobre todo el putamen, con hipointensidad en secuencias T2 y aumento de señal periférico. Hay una atrofia selectiva de la protuberancia con aplanamiento inferior de ésta, de los pedúnculos cerebelosos medios, cerebelo (mayor cortical que en el vermis) y oliva inferior. Es característica una alteración de señal en la protuberancia, en forma de cruz (referido como “hot cross bun sign” en la bibliografía inglesa), y en las vías pontocerebelosas con hiperintensidades en los pedúnculos cerebelosos medios, donde se ha descrito asimismo una alteración de difusión33–37 (fig. 7).
 Corte sagital potenciado en T1 en el que se observa una importante atrofia de la protuberancia con aplanamiento inferior. b) Corte axial potenciado en T2 donde además de la atrofia pontina se observa una alteración de señal en forma de cruz. También se demuestra una atrofia, con hiperintensidad de señal, en los pedúnculos cerebelosos medios.")
Atrofia multisistema. Resonancia magnética premortem. a) Corte sagital potenciado en T1 en el que se observa una importante atrofia de la protuberancia con aplanamiento inferior. b) Corte axial potenciado en T2 donde además de la atrofia pontina se observa una alteración de señal en forma de cruz. También se demuestra una atrofia, con hiperintensidad de señal, en los pedúnculos cerebelosos medios.
La enfermedad de Hallervorden-Spatz también presenta cuerpos de Lewy con α-sinucleína y depósitos de hierro en núcleos extrapiramidales, sobre todo en el globo pálido y la sustancia negra, así como distrofia neuroaxonal con gliosis. Clínicamente se caracteriza por un cuadro parkinsoniano y deterioro mental. El 50% de los casos son familiares y se ha demostrado una herencia autosómica recesiva. En RM se ha descrito una hipointensidad, con hiperintensidad central en las secuencias T2 en los núcleos extrapiramidales, sobre todo en el globo pálido, con imagen en “ojos de tigre”, típica aunque no específica de esta enfermedad38.
Degeneración lobular frontotemporalLa degeneración lobular frontotemporal (DLFT) es un grupo heterogéneo de trastornos que asocian una degeneración de los lóbulos frontales y temporales en su porción anterior. Constituye entre el 5 y el 20% de las demencias primarias aunque son más frecuentes en el grupo de inicio temprano. Presenta una tasa más rápida de deterioro.
Clínicamente se caracteriza por cambios de personalidad y carácter, con deterioro progresivo de las funciones ejecutivas y del lenguaje. La memoria y habilidades visuoespaciales están preservadas hasta fases tardías. Desde un punto de vista clínico, inicialmente, se clasifican en demencias frontotemporales si dominan las alteraciones de conducta y personalidad (dentro de las que se incluirían como un subgrupo la enfermedad de Pick), y afasia progresiva primaria si predomina la alteración del lenguaje, que puede ser no fluente en la afasia progresiva no fluente y fluente en la afasia semántica. Con la evolución de la enfermedad estas 3 formas clínicas se solapan y desarrollan un cuadro de mutismo y alteración de conducta7.
En radiología se puede observar una atrofia selectiva frontotemporal que puede ser simétrica o asimétrica. En el lóbulo temporal la afectación predomina en la región anterior, con relativa preservación de la región medial. En la RM se han descrito alteraciones de señal en la corteza y la sustancia blanca. La PET y la SPECT pueden demostrar una alteración metabólica y vascular frontotemporal1,39,40.
En anatomía patológica se observan atrofia, espongiosis cortical con pérdida neuronal y gliosis. La clasificación de este grupo de enfermedades ha presentado importantes cambios debido a los avances en inmunohistoquímica y biología molecular. Las formas patológicas de DLFT no predicen de forma fiable los distintos fenotipos clínicos de presentación de estas enfermedades.
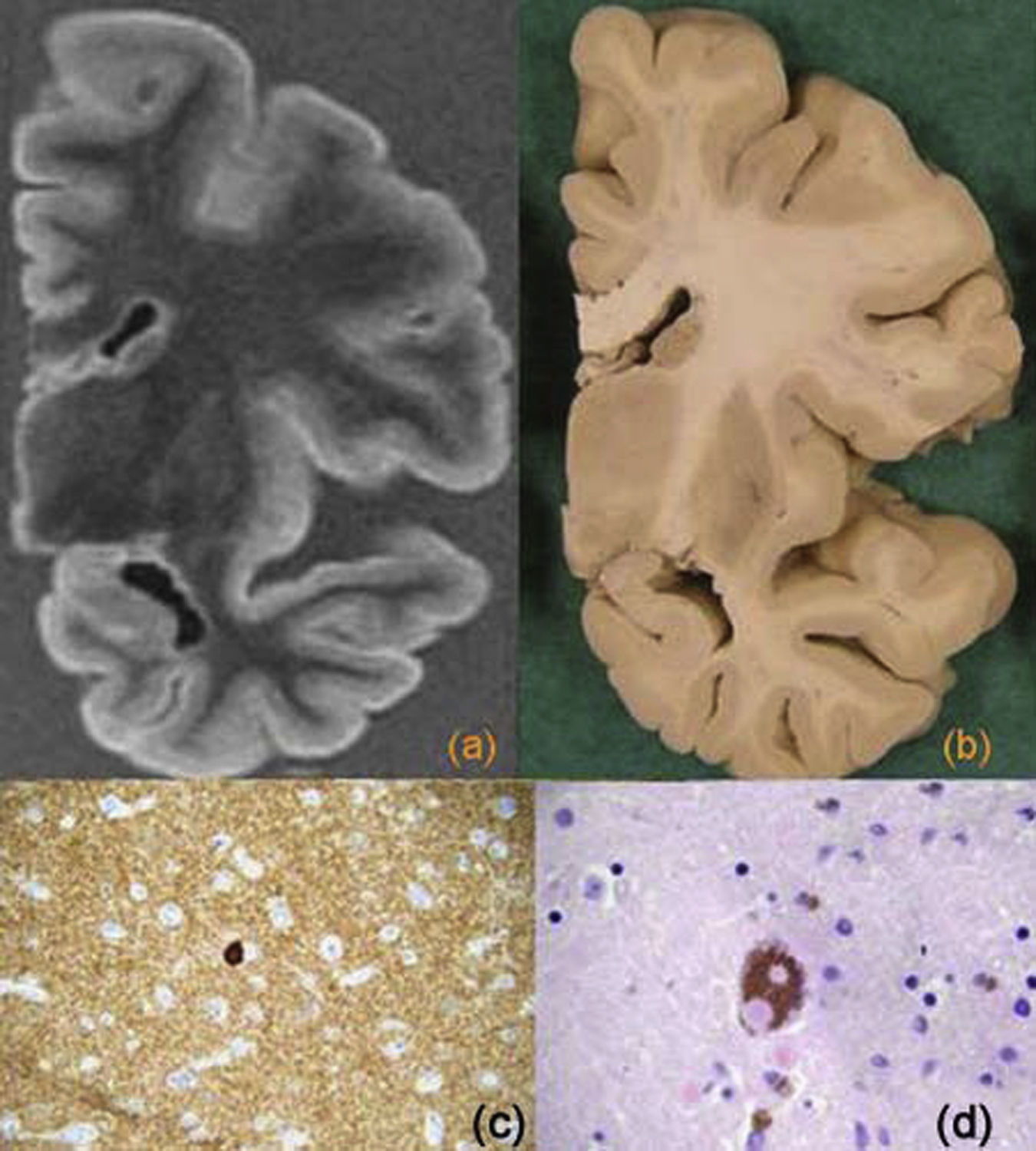
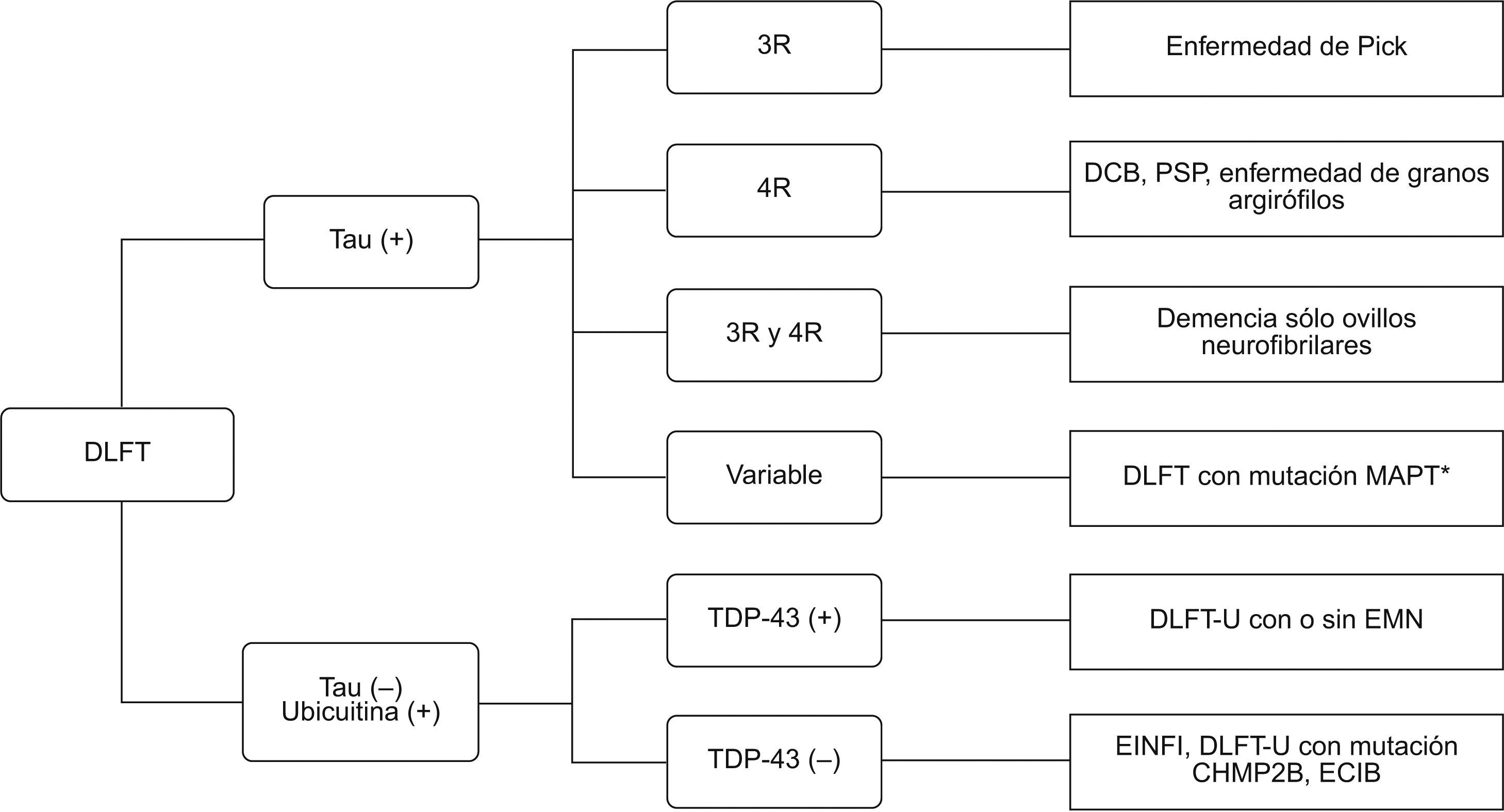
Recientemente se han propuesto unos criterios de consenso para el diagnóstico patológico que se explicarán brevemente41 (fig. 8). Se mantiene la división inicial de este grupo de enfermedades según su positividad a la proteína tau. Así, dentro del grupo de las taupatías se incluyen varios procesos que se subdividen según la isoforma de la proteína tau presente. La DLFT con cuerpos de Pick se caracteriza por la presencia de la forma tau 3R, mientras que en la degeneración corticobasal, la parálisis supranuclear progresiva y la enfermedad de granos argirófilos predominan las formas 4R, en la demencia con ovillos neurofibrilares se observan formas mixtas 3R y 4R y la DLFT con mutación del gen MAPT es heterogénea en las formas tau. El segundo grupo lo constituyen las formas de DLFT con inclusiones tau (−), ubicuitina (+) y α-sinucleína (−), también conocido como DLFT-U (estas enfermedades pueden asociar o no enfermedad de motoneurona). La proteína TDP-43 se ha identificado como el principal componente de estas inclusiones y define ahora un nuevo grupo de enfermedades llamado TDP-43 proteinopatías, que constituye el hallazgo histológico más frecuente en pacientes con DLFT (fig. 9).
. DCB: degeneración corticobasal; DLFT-U: DLFT-ubicuitin (+); ECIB: enfermedad con cuerpos de inclusión basódilos; EINFI: enfermedad con inclusiones intraneuronales de filamentos intermedios; EMN: enfermedad de motoneurona; PSP: parálisis supranuclear progresiva. *También conocida con DFT con parkinsonismo ligado al cromosoma 17.")
Clasificación de degeneración lobular frontotemporal (DLFT). DCB: degeneración corticobasal; DLFT-U: DLFT-ubicuitin (+); ECIB: enfermedad con cuerpos de inclusión basódilos; EINFI: enfermedad con inclusiones intraneuronales de filamentos intermedios; EMN: enfermedad de motoneurona; PSP: parálisis supranuclear progresiva. *También conocida con DFT con parkinsonismo ligado al cromosoma 17.
. Corte coronal de resonancia magnética (a) y pieza macroscópica (b) con una importante atrofia cortical frontal y una alteración de señal en la sustancia blanca. c) Imagen microscópica con técnica de inmunohistoquímica anti TDP-43 que muestra una inclusión intranuclear.")
Degeneración lobular frontotemporal TDP-43 (+). Corte coronal de resonancia magnética (a) y pieza macroscópica (b) con una importante atrofia cortical frontal y una alteración de señal en la sustancia blanca. c) Imagen microscópica con técnica de inmunohistoquímica anti TDP-43 que muestra una inclusión intranuclear.
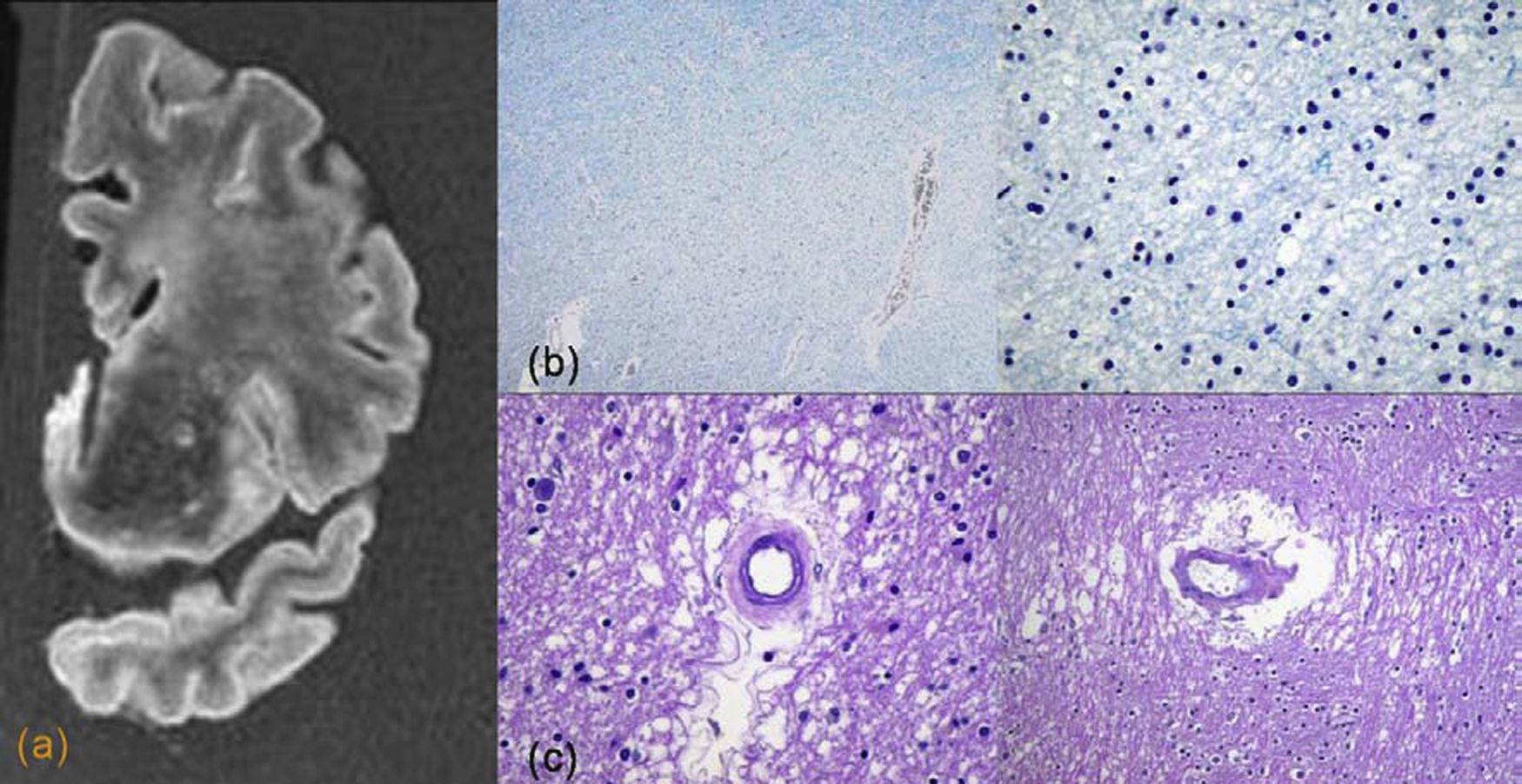
La enfermedad de Pick se considera un prototipo del grupo de demencias frontotemporales, aunque únicamente constituye el 15% de éstas. Se caracteriza por una atrofia importante, sobre todo de la corteza prefrontal y del tercio anterior del giro temporal superior, con circunvoluciones en “filo de cuchillo”. También hay una atrofia del núcleo estriado. La sustancia blanca presenta una consistencia gelatinosa por desaparición de la mielina secundaria a la pérdida neuronal y gliosis. Se caracteriza por la presencia de neuronas balonizadas o células de Pick y de cuerpos de Pick tau (+), en la corteza e hipocampo7,33,42 (fig. 10).
 Resonancia magnética con corte sagital potenciado en T1 y corte coronal FLAIR. b) Hemisferio cerebral, visión lateral y corte coronal. Hay una importante atrofia de la corteza frontal y temporal anterior con alteración de señal en la sustancia blanca frontal. Imágenes microscópicas de la corteza cingular con tinción HE (c) y tau (d), en las que se observan células balonizadas y cuerpos de Pick intraneuronales tau (+).")
Enfermedad de Pick. a) Resonancia magnética con corte sagital potenciado en T1 y corte coronal FLAIR. b) Hemisferio cerebral, visión lateral y corte coronal. Hay una importante atrofia de la corteza frontal y temporal anterior con alteración de señal en la sustancia blanca frontal. Imágenes microscópicas de la corteza cingular con tinción HE (c) y tau (d), en las que se observan células balonizadas y cuerpos de Pick intraneuronales tau (+).
La degeneración corticobasal es una entidad clinicopatológica heterogénea que asocia degeneración en áreas corticales, ganglios basales y sustancia negra. Los pacientes presentan síntomas parkinsonianos atípicos. Otra forma de presentación es la de una apraxia progresiva con presencia de “mano alienígena”. Se desarrolla una alteración motora por afectación de la vía piramidal. Algunos pacientes asocian demencia de tipo frontotemporal. Se puede observar una atrofia, sobre todo hemisférica izquierda, con aumento de tamaño de los surcos pre y poscentral, y de la región perisilviana. En anatomía patológica hay pérdida neuronal y gliosis con neuronas balonizadas en la corteza premotora y cingulada. En la sustancia negra se observa una pérdida neuronal, gliosis y ovillos neurofibrilares. Un hallazgo característico son las placas astrocitarias7,33,43.
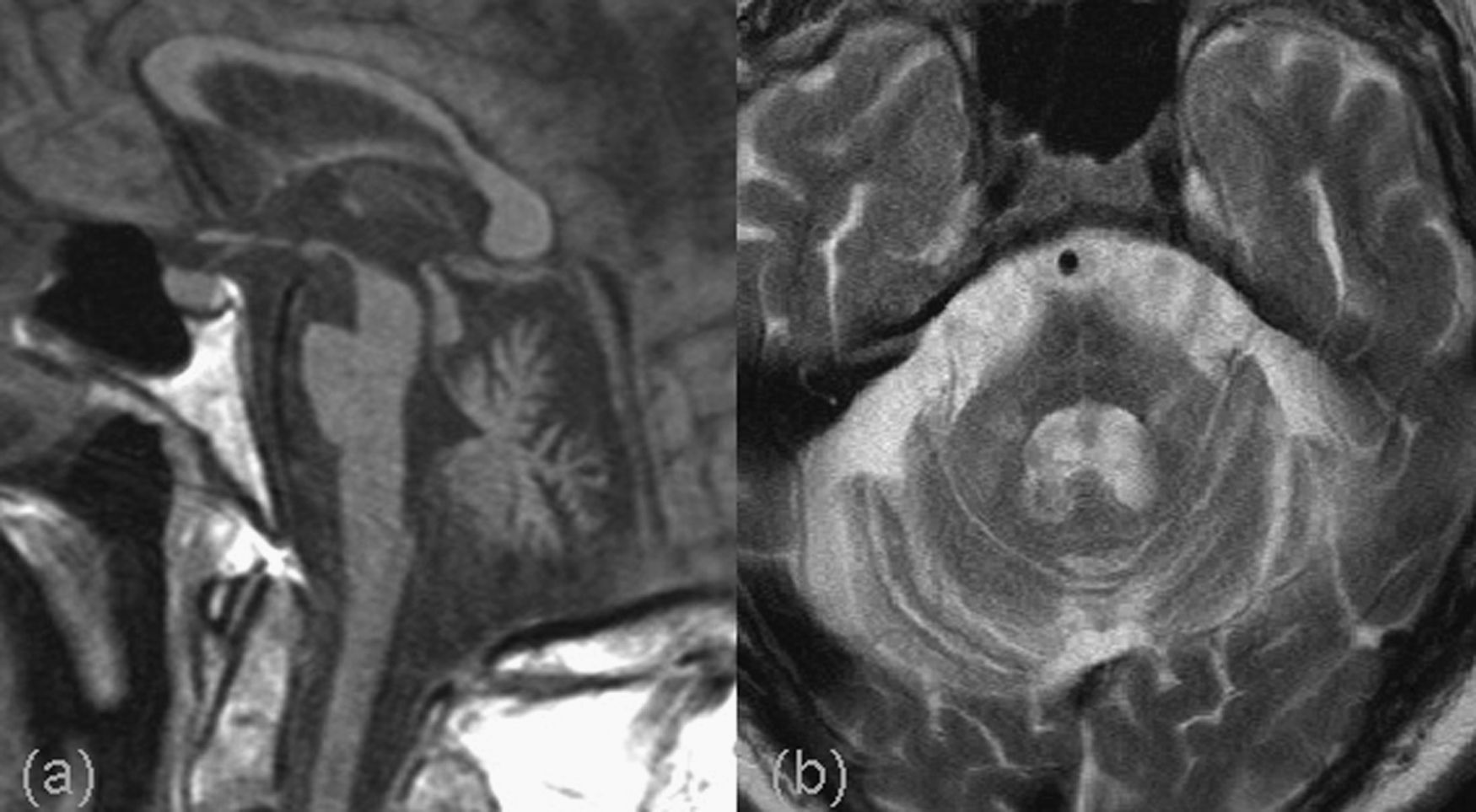
La parálisis supranuclear progresiva es una enfermedad que se caracteriza por parkinsonismo y oftalmoplejía supranuclear. También se puede desarrollar una parálisis seudobulbar y a veces demencia. Se afectan especialmente el mesencéfalo y el puente, aunque también el globo pálido y el núcleo subtalámico, y se observa una disminución de la población neuronal y gliosis. El acúmulo de proteína tau adopta varias formas: en ovillos neurofibrilares de apariencia globosa en neuronas, astrocitos en “penacho” (característicos de esta enfermedad) y “espinosos”, y en el interior de oligodendrocitos en los llamados cuerpos espirales. En radiología se observa una atrofia focal y/o alteración de señal en el tronco cerebral, especialmente en el mesencéfalo, con mayor afectación de los tubérculos cuadrigéminos y pedúnculos cerebelosos superiores. Se ha descrito como característico el signo del “colibrí” o “pingüino” en los cortes sagitales de RM. También puede haber una hiperintensidad en la región periacueductal, una disminución de la pars compacta de la sustancia negra y una disminución de señal en el putamen33,35,36,44 (fig. 11).
 Resonancia magnética (RM) premortem, corte sagital potenciado en T1 donde se observa una atrofia selectiva del mesencéfalo (signo del colibrí). RM posmortem, corte coronal FLAIR (b) y corte coronal macroscópico (c) que muestran un cerebro normal. Imagen microscópica teñida con tau donde se observa un ovillo neurofibrilar de forma globosa y un astrocito con aspecto en penacho (d).")
Parálisis supranuclear progresiva. a) Resonancia magnética (RM) premortem, corte sagital potenciado en T1 donde se observa una atrofia selectiva del mesencéfalo (signo del colibrí). RM posmortem, corte coronal FLAIR (b) y corte coronal macroscópico (c) que muestran un cerebro normal. Imagen microscópica teñida con tau donde se observa un ovillo neurofibrilar de forma globosa y un astrocito con aspecto en penacho (d).
La esclerosis lateral amiotrófica (ELA) es el prototipo de enfermedad de neurona motora con pérdida muscular. Aparecen déficit cognitivos leves en el 30–40% de los pacientes, pero sólo aproximadamente el 5% presenta una demencia franca, de tipo frontotemporal. En estos pacientes, además de los hallazgos patológicos característicos de la ELA hay una pérdida neuronal y gliosis en la corteza, los ganglios basales y la sustancia negra, con presencia de inclusiones ubicutina (+), tau (−) en corteza e hipocampo. Estas inclusiones que definen el grupo DLFT-U, tal como se ha descrito anteriormente, pueden estar presentes en pacientes con o sin enfermedad de motoneurona7,33,41.
OtrasAdemás de los grupos principales que se han descrito, hay un importante número de procesos degenerativos, menos frecuentes, que pueden asociar cuadros de demencia y de los que no podemos realizar, en este trabajo, una revisión exhaustiva. Dentro de éstos se podrían incluir la corea de Huntington, las enfermedades por priones, la esclerosis del hipocampo, las alteraciones genéticas, como la lipofuscinosis ceroide (enfermedad de Kufs) o las encefalopatías mitocondriales, etc. De todos estos procesos nos referiremos brevemente a algunos de ellos.
La corea de Huntington es una enfermedad hereditaria autosómica dominante, que se desarrolla en la cuarta o quinta década por mutación del gen que codifica la proteína huntingtina (cromosoma 4). Se caracteriza por un cuadro de coreoatetosis y demencia. Se puede observar una atrofia del núcleo caudado y putamen. La atrofia de la cabeza del núcleo caudado provoca una alteración morfológica característica de las astas frontales. Además se ha descrito un aumento y una disminución de la señal en T2 en el núcleo estriado. La histología muestra pérdida neuronal y gliosis en el núcleo estriado y cuerpos de inclusión intranucleares por acúmulo de huntingtina, más frecuentes en el córtex45 (fig. 12).
 y macroscópica (b), en los que se ve una importante atrofia de la cabeza del núcleo caudado. c) Imagen microscópica del núcleo caudado, teñida con ubicuitina, donde se observa una inclusión intranuclear (tinción antiubicuitina).")
Corea de Huntington. Cortes coronales de resonancia magnética (a) y macroscópica (b), en los que se ve una importante atrofia de la cabeza del núcleo caudado. c) Imagen microscópica del núcleo caudado, teñida con ubicuitina, donde se observa una inclusión intranuclear (tinción antiubicuitina).
La enfermedad de Creutzfeldt-Jakob (ECJ) es la enfermedad priónica que en la actualidad se asocia con mayor frecuencia a demencia. Aunque su incidencia es muy baja, su diagnóstico puede tener implicaciones de salud pública importantes. Hay 4 formas: esporádica, iatrogénica, familiar y variante. La mayoría de los casos son esporádicos. Clínicamente se trata de una demencia rápidamente progresiva, que suele acompañarse de mioclonías, signos extrapiramidales, ataxia cerebelosa y ceguera cortical. En el EEG se pueden ver complejos periódicos trifásicos característicos. El agente transmisible de la enfermedad es una isoforma anormal de una proteína priónica humana, codificada en el brazo corto del cromosoma20. En patología se ven cambios espongiformes, pérdida neuronal y astrocitaria. En la RM es característico un aumento de señal bilateral, simétrico o asimétrico, en el caudado y putamen, en las imágenes potenciadas en T2. También se ha descrito un aumento de señal en la corteza. Estas alteraciones se acompañan de restricción de difusión en fases iniciales. Al progresar la enfermedad se atrofian el estriado y neocórtex. En la variante de ECJ la anomalía descrita con mayor frecuencia es la alteración de señal en el pulvinar talámico. Las formas familiares se presentan con un amplio rango de fenotipos clinicopatológicos. En éstos se incluyen la enfermedad de Gerstmann-Straussler-Scheinker, la ECJ familiar, el insomnio familiar fatal y otros1,33.
La lipofuscinosis ceroide es una enfermedad producida por el almacenamiento excesivo de lipopigmentos en el citoplasma neuronal y posiblemente glial. Hay una forma clínica en niños y otra juvenil y del adulto o enfermedad de Kuf. En esta última se presentan convulsiones tonicoclónicas y demencia. Se han identificado 7 genes y descrito aproximadamente 150 mutaciones. Se puede observar una atrofia importante con afectación secundaria de sustancia blanca. Hay neuronas balonizadas con acúmulo de lipopigmentos, PAS (+), en córtex, cerebelo, núcleos subcorticales y tronco cerebral46 (fig. 13).
 y macroscópicos (b) con una importante atrofia global y alteración de señal de la sustancia blanca. c) Imagen microscópica con tinción PAS, donde se observan acúmulos de lipofuscina en corteza.")
El síndrome del cromosoma X frágil es la causa hereditaria más frecuente de retraso mental en varones. Hay una forma de premutación de este cromosoma no asociada a retraso mental, dentro de la cual se ha descrito un subgrupo de pacientes varones mayores, que presenta un cuadro de ataxia cerebelosa y temblor que se acompaña de deterioro cognitivo. En el estudio patológico se han encontrado inclusiones intranucleares eosinofílicas en neuronas y astrocitos, en el córtex, en el tronco cerebral y en el hipocampo, así como espongiosis en la sustancia blanca cerebelosa. En la RM se han descrito signos de atrofia y alteraciones de señal, hiperintensa en las secuencias potenciadas en T2, en la sustancia blanca cerebelosa y en los pedúnculos cerebelosos medios47.
ConclusiónLas enfermedades degenerativas constituyen un grupo heterogéneo que incluye procesos poco frecuentes y enfermedades muy prevalentes, como la EA.
La radiología tiene un papel importante en el diagnóstico y en el control evolutivo de los pacientes con alteraciones cognitivas, así como en los ensayos clínicos de los nuevos tratamientos que se están desarrollando. Aunque clásicamente se han considerado como poco específicas, las pruebas de imagen pueden aportar, tal como se ha descrito, datos en la distribución de la atrofia (el hipocampo en la EA o la atrofia frontotemporal en la DLFT), imágenes características como la atrofia del mesencéfalo en la parálisis supranuclear progresiva o de la protuberancia en la atrofia multisistémica e incluso criterios diagnósticos en la demencia vascular. El conocimiento de estos procesos, junto con una colaboración más estrecha con los clínicos implicados, permitirá obtener un mejor rendimiento diagnóstico de las exploraciones radiológicas y colaborar en la atención de estos pacientes.
Declaración de conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

