


Apoptosis es el nombre que dio Kerr1 a los cambios morfológicos específicos que sufren las células y que culminan en la muerte celular por un mecanismo diferente de la necrosis. Este concepto, propuesto hace más de cien años por Vogt (1842), se forjó al concretarse que la muerte celular no es un proceso aleatorizado y que tiene unas características morfológicas peculiares y específicas2.
Las células apoptóticas desarrollan condensación y segmentación del núcleo, condensación y fragmentación del citoplasma y a menudo activación de endonucleasas que provocan una extensa fragmentación por proteolisis del ADN cromosómico con formación de oligofragmentos ADN, denominados nucleosomas o cuerpos apoptóticos3.
Los estímulos celulares que pueden desencadenar apoptosis son diversos: hipoxia e isquemia, exposición a productos tóxicos, traumatismo y supresión de factores de crecimiento. Muchas veces este proceso se desencadena por supresión de señales vitales que al fallar permiten que se active un programa intrínseco de muerte celular4.
Para ello ha sido fundamental el descubrimiento de diferentes receptores presentes en numerosas células, y su potencial activación por proteínas ligadas a dichos receptores (Fas, FasL, enzima b-conversor de la interleucina-1), así como el reconocimiento de genes que regulan dicha apoptosis (proteína CED-3, proteína P53)5.
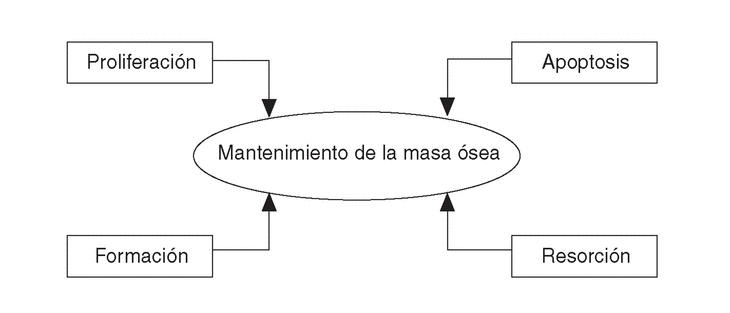
El papel de la apoptosis de las células óseas en la patogénesis de la osteoporosis ha complicado el inicial concepto de remodelado a base de formación-resorción, al introducir otro binomio: proliferación celular-apoptosis6 (fig. 1).
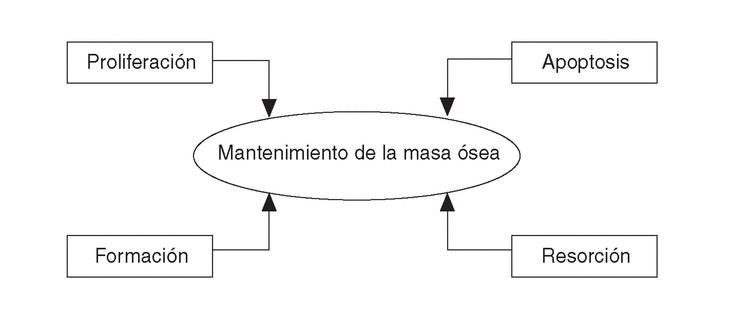
Figura 1. Nuevo concepto de remodelado óseo.
El remodelado óseo se realiza por una acción combinante entre los osteoclastos y los osteoblastos, estructura anatómica conocida como unidad multicelular básica (BMU). Durante la vida media de una BMU, entre seis y nueve meses, los osteoclastos y osteoblastos requieren un reemplazamiento continuo en función de las características, localización y función del hueso7.
La formación del osteoblasto se realiza a partir de las células mesenquimatosas pluripotenciales, muchas de ellas necesarias para crear la hematopoyesis, mientras que los precursores del osteoclasto pertenecen a la línea monocito-macrófago. El control del crecimiento y formación de estas células se realiza por factores de crecimiento y citocinas producidas en la médula ósea, a su vez moduladas por hormonas sistémicas8.
La puesta en marcha de estos precursores mesenquimales se realiza por las proteínas morfogenéticas óseas (BMP), miembros de la superfamilia factor de crecimiento transformante-B (TGF-B); la misma familia de polipéptidos que controla la formación de hueso endocondral durante la embriogénesis a la vez que puede inducir formación ósea ectópica9. También conocemos las fases modulares de la osteoclastogénesis sobre los precursores de osteoblastos, a través de citocinas como el factor de necrosis tumoral RANK, también conocido como ligando de osteoprotegerina10.
Los osteoclastos tras la reabsorción ósea mueren por apoptosis y son removidos por los fagocitos. Los osteoblastos, con una vida media más larga, en parte se transforman en osteocitos, pero el 65% muere por apoptosis durante su misión de recomponer la cavidad erosionada por los osteoclastos. Es el balance entre proliferación celular y apoptosis lo que determina la prevalencia de osteoblastos y osteoclastos, de aquí que los cambios en la apoptosis significan cambios entre la formación y resorción del hueso11.
Sólo recientemente conocemos el papel de los osteocitos, el mayor número de células en el hueso. Los osteocitos preferentemente modulan las señales que provienen de los estímulos mecánicos. Utilizan señales moleculares similares a otros tejidos, como la generación de óxido nítrico y las prostaglandinas, así como las uniones intercelulares comunicantes (gap junctions). También movilizan el hueso destruído a través de mecanismos ligados a su propia apoptosis, por la secreción de proteínas especializadas, como la osteopontina. Al poseer receptores para la PH, PrPTH y para los estrógenos regulan el remodelado óseo a través de la apoptosis12.
No es raro que hoy en día se intente explicar por un mecanismo apoptótico la patogenia de osteoporosis. En la osteoporosis tipo I o postmenopáusica se conoce el papel de la deficiencia estrogénica, que resulta en un aumento del número y actividad de los osteoclastos, lo que contribuye a un aumento de la resorción. Al mismo tiempo aumenta la apoptosis de los osteoblastos, lo que contribuye al desequilibrio entre resorción y formación ósea13.
Menos se conoce sobre los efectos del envejecimiento y la apoptosis de osteoblastos y osteoclastos para explicar la osteoporosis tipo II o senil. Aquí desempeña un papel más importante la muerte de los osteocitos que conlleva un defecto en la reparación del daño y de la fuerza óseos14.
La apoptosis explica la osteoporosis inducida por corticoides, ya que aumenta la apoptosis de los osteocitos, al tiempo que estimula la de los osteoblastos y frena la de los osteoclastos. A través de este mecanismo también puede explicarse un mayor incremento de la necrosis vascular ósea y la presencia de osteoporosis tras la supresión del tratamiento con esteroides15.
Nuestros conocimientos también ayudan a explicar el significado del tratamiento de la osteoporosis. Sabemos que diferentes fármacos ejercen un efecto alterando la acción de la apoptosis y por tanto el destino de los osteoclastos y osteoblastos16.
Los estrógenos y el raloxifeno estimulan la apoptosis de los osteoclastos y por lo tanto previenen la pérdida ósea16. Los aminobifosfonatos, la calcitonina y la vitamina K actúan también promoviendo la apoptosis de los osteoclastos17-19.
Otros fármacos disminuyen la prevalencia de apoptosis de los osteoblastos explicando el estímulo de la formación de hueso, y por lo tanto el aumento de la densidad ósea. Entre éstos, el mejor estudiado es la parathormona, aunque posiblemente también los bifosfonatos y la calcitonina en concentraciones nanomolares18,20.
Estos hallazgos abren un esperanzador presente a nuestros conceptos de osteoporosis, y marcan objetivamente los efectos de los agentes terapéuticos que al corregir el desequilibrio en el número y calidad de la células del BMU aumentan la masa ósea, y por lo tanto, sientan las bases celulares para disminuir el riesgo de fracturas en la osteoporosis.
NOTICIAS
LA FUNDACIÓN HISPANA DE OSTEOPOROSIS Y ENFERMEDADES METABÓLICAS ÓSEAS (FHOEMO)
Convoca DOS BECAS FHOEMO PARA JÓVENES INVESTIGADORES 2001 de acuerdo con las siguientes
BASES
1. La Beca permitirá la realización de un período de aprendizaje y formación, sobre aspectos experimentales y/o clínicos en el campo de la Osteoporosis y Enfermedades Metabólicas Óseas, en un centro clínico especializado de ámbito nacional.
2. Las Becas estarán dotadas con 400.000 pesetas.
3. Los candidatos serán posgraduados, menores de 40 años, interesados en el estudio de estas enfermedades.
4. La fecha límite de recepción será el 1 de junio de 2001.
5. La Beca será concedida por un Jurado que estará compuesto por cuatro miembros del Comité Científico de la FHOEMO, actuando como secretaria del Jurado, con voz pero sin voto, la Secretaria de la Fundación.
6. La propuesta consistirá en una concisa explicación sobre el proyecto, el Curriculum Vitae del solicitante y la aceptación del jefe de Servicio donde vaya a desarrollar su labor.
7. La documentación se remitirá a la Secretaría de la FHOEMO (Gil de Santivañes, 6, 2.o D. 28001 Madrid. Tel/Fax: 91 578 35 10).
8. La decisión del Jurado será inapelable y las Becas podrán ser declaradas desiertas si, a juicio del jurado, se estimase que los proyectos presentados no reúnen los méritos suficientes.
9. Los aspirantes aceptarán las presentes bases por el mero hecho de concurrir a esta convocatoria.