


effect.
INTRODUCCION
Si bien es conocido que los esteroides sexuales ejercen un papel regulador sobre el metabolismo óseo, en los últimos años se ha comenzado a especular sobre la dependencia del sexo en este efecto.
Los receptores de esteroides sexuales son factores de transcripción citonucleares cuyas estructuras son modificadas por medio de la unión con el ligando. Aparte del ya conocido proceso del receptor de hormonas sexuales, que una vez modificado por su ligando actúa como promotor de genes, otros procesos que modulan los efectos de las hormonas sexuales en las células diana son:
1. Diversos mediadores moleculares que actúan de forma autocrina y paracrina.
2. El papel que las fuerzas biomecánicas desempeñan en el nivel de masa ósea.
3. Los efectos no genómicos y no dependientes del sexo, recientemente descubiertos, de estrógenos (E) y andrógenos (A).
En este artículo se abordan estos y otros hallazgos moleculares que tratan de explicar las acciones fisiológicas de las hormonas esteroideas sobre el hueso.
Por otra parte se revisarán los datos obtenidos de las investigaciones in vitro e in vivo en animales y humanos sobre el modo de acción de los A (directo e indirecto) sobre el hueso y su importancia en la homeostasis ósea y en la protección frente a la osteoporosis.
SINTESIS Y METABOLISMO DE LOS ESTEROIDES SEXUALES
En mujeres premenopáusicas más del 95% del estradiol (E2) y la mayor parte de estrona (E1) proceden de la secreción ovárica. El resto de E circulantes en la mujer premenopáusica y la mayor parte de ellos en la mujer posmenopáusica proceden de la conversión periférica de precursores de esteroides. En varones más del 95% de la testosterona (T) procede de la secreción testicular. La T sérica en mujeres premenopáusicas procede un 25% de secreción ovárica, un 25% de secreción adrenal y un 50% de conversión periférica. En la mujer posmenopáusica las fuentes son similares, excepto la secreción ovárica de T, que disminuye. En la mayoría de los tejidos diana, la 5*-dihidrotestosterona (DHT), formada a partir de T por la acción de la enzima 5*-reductasa, es la principal fuente de actividad androgénica. El córtex adrenal, y en menor medida las gónadas, secretan grandes cantidades de A C19: dehidroepiandrostendiona (DHEA), DHEA sulfato (DHEA-S) y 4-androstenediona. Aunque se trata de A débiles por sí mismos, son una importante fuente para la síntesis extragonadal de potentes esteroides sexuales, ya que gracias a la aromatasa se convierten en E1 y gracias a la esteroide sulfatasa, la 17-ß-dihidroxiesteroidedeshidrogenasa y la 3ß-hidroxies
teroidedeshidrogenasa en T.
La biosíntesis extragonadal desempeña un importante papel. Muchos tejidos periféricos, incluyendo el hueso, pueden sintetizar E1 a partir de C19 esteroides. E2 y DHT pueden ser sintetizados directamente desde T. El principal lugar de conversión es el tejido adiposo. La biosíntesis extragonadal se incrementa en personas obesas y en mujeres posmenopáusicas1.
Labrie et al2 llamaron «acción autocrina» al proceso por el que los esteroides son sintetizados por una célula diana y su acción se lleva a cabo sin ser liberado al fluido extracelular. Los tejidos extragonadales que sintetizan E1 y E2 utilizan las mismas vías enzimáticas que las empleadas en la síntesis gonadal, con la excepción de que no pueden sintetizar C19 esteroides y dependen de precursores circulantes por tanto. Las enzimas clave involucradas en estos procesos se encuentran presentes en osteoblastos. La enzima aromatasa también se expresa en los condrocitos3. De los esteroides sexuales activos en tejidos periféricos, el 50% del total de los A en el varón adulto, el 75% de los E en mujeres premenopáusicas y casi todos los E en mujeres posmenopáusicas proceden de esta síntesis extragonadal.
La biosíntesis de esteroides sexuales es en gran medida tejido-específica: en las gónadas el principal E sintetizado es E2, en el tejido adiposo es E1 y en la placenta es estriol. La expresión del gen de la aromatasa en estos lugares se halla bajo control de promotores tejido-específicos que son regulados por diferentes factores de transcripción y citocinas. Existen pruebas de que la producción extragonadal de E es un proceso regulado. Se ha demostrado que la interleucina -6(IL-6) regula la actividad de 17ß-HSD en células de cáncer de mama4 y que la IL-1ß y el factor de necrosis tumoral * (TNF*) regulan la actividad de CYP19 aromatasa en osteoblastos1. La producción de estas mismas citocinas proinflamatorias en el microambiente del hueso se incrementa por el déficit de E. Finalmente, Eyre et al5 demostraron que la línea celular osteoblástica en ratas, ROS 17/2,8, podía sintetizar E1, E2 y T a partir de 4-androstenediona y que esta síntesis podía ser favorecida por 1,25-dihidroxivitamina D y dificultada por glucocorticoides.
MECANISMO DE ACCION DE LOS ESTEROIDES SEXUALES SOBRE EL HUESO
MECANISMO GENOMICO
Receptores esteroideos
Los receptores esteroideos pertenecen a la familia de los «receptores nucleares», en la cual se incluyen moléculas que ligan además de hormonas esteroideas vitamina A, ácido retinoico y hormonas tiroideas.
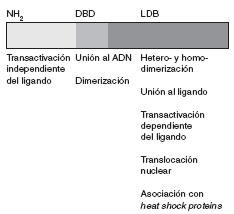
La secuencia de aminoácidos del receptor para esteroides ha sido plenamente identificada. Se trata de una proteína de 777 aminoácidos, organizada en varios dominios con funciones específicas (fig. 1).
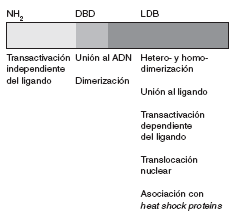
Fig 1. Representación esquemática del receptor esteroideo
La región aminoterminal (AF-1) participa en la transactivación a través de la cual se promueve la transcripción génica. Adyacente a esta región se encuentra una región básica que contiene dos dedos de zinc que se unen al ADN. La secuencia de aminoácidos localizada entre ambos dedos es la responsable de establecer contactos específicos con secuencias de ADN. El segundo dedo de zinc estabiliza la unión y aumenta la afinidad del receptor por estas secuencias. El último dominio es la región carboxiterminal, que es responsable de la unión al ligando, la dimerización o heterodimerización del receptor y la asociación con heat shock proteins. Asimismo, también contribuye a la transactivación dependiente del ligando (función situada en un subdominio llamado AF-2), que se relaciona con la actividad transcripcional.
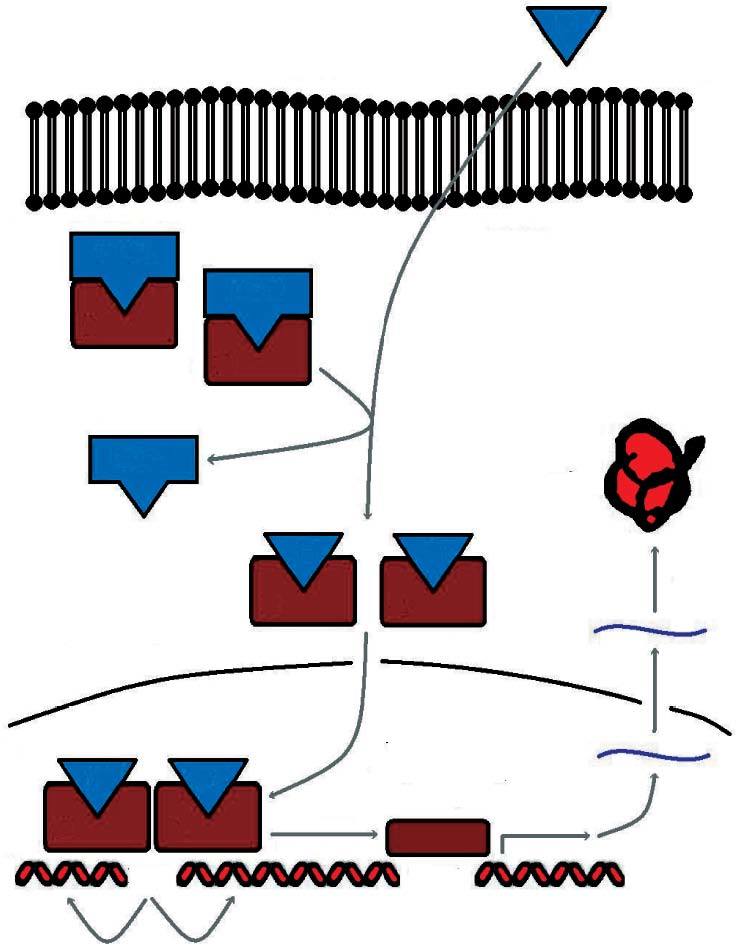
En condiciones basales se encuentran situados en el citoplasma formando complejos multiméricos con las heat shock proteins (hsp 90, hsp 70, hsp 56). Cuando se une la hormona al receptor se disocian estos complejos y el receptor inicia su traslocación al núcleo, donde se une a secuencias específicas del ADN denominadas elementos de respuesta a las hormonas esteroideas (ERE) (fig. 2).
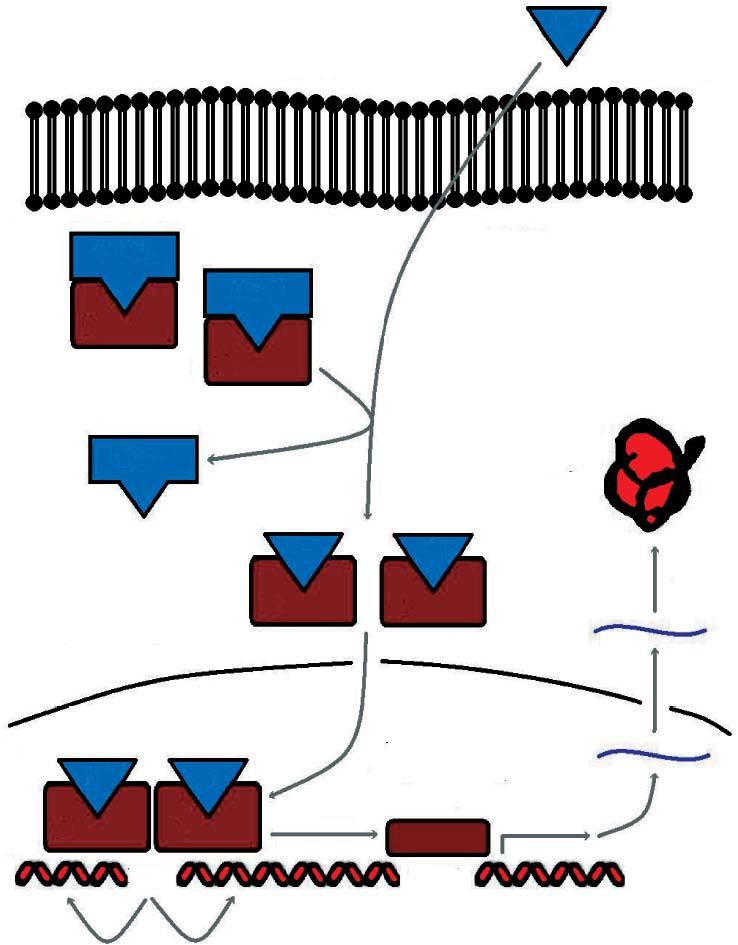
Fig 2. Sistema de señalización del receptor esteroideo
Esta actividad transcripcional se encuentra modulada en los tejidos diana por factores correguladores. Así, hay factores que aumentan la transcripción (coactivadores) y factores que la disminuyen (cosupresores).
Transducción por medio de receptores esteroideos en hueso y cartílago
Hasta 1988 se pensaba que los esteroides sexuales afectaban al esqueleto sólo de forma indirecta, regulando la secreción de hormonas calciotrópicas. Sin embargo, el hallazgo de receptores esteroideos en las distintas células óseas, gracias al desarrollo de técnicas de investigación más sensibles, puso de manifiesto la existencia de una acción directa de estas hormonas sobre el hueso. Así, en 1986 fue clonado el receptor estrogénico alfa (RE*); en 1988 el receptor androgénico (RA)6,7 y finalmente en 1995 un segundo receptor estrogénico llamado REß8. También han sido descritos heterodímeros RE*/REß.
En el cartílago de crecimiento humano in vivo se detectan RA en todas sus capas tanto inmunohistoquímicamente9-13 como por técnicas de hibridación in situ13 sin diferencias dependientes del sexo9,10,14. Asimismo, también están presentes en todas las capas los RE*10,12,15,16 existiendo controversia sobre la localización de REß, pues mientras unos estudios los localizan sólo en los condrocitos hipertróficos17, otros los sitúan en todas las capas12,15,18.
La expresión de RA en cultivo de osteoblastos fue descrita por primera vez en 1989 por Colvard et al19, y posteriormente múltiples estudios han confirmado la expresión de ARNmRA en osteoblastos y osteocitos9,11,13,19-22. La expresión de RA es mayor en OB del hueso cortical que en los del hueso trabecular, mientras que no existen diferencias significativas en su cantidad dependientes del sexo23. Recientemente se ha comprobado que los A regulan a la alta la expresión de su propio receptor en los osteoblastos24-27.
Los RE se describieron por primera vez en cultivos de osteoblastos humanos en 198828,29, pero aún hoy no existe consenso sobre su expresión relativa durante el proceso de diferenciación y en cuanto a su localización dentro del esqueleto. Así, parece que RE* predomina en el hueso cortical, mientras que REß lo hace en el hueso trabecular15.
En osteoclastos de modelos animales se han hallado RA30,31 tanto in vivo como in vitro, no así en OC humanos in vivo9,11, lo que llevó a pensar que la acción de los A sobre los OC se realizaba de forma indirecta a través de los OB32. No obstante, recientes estudios in vitro han demostrado que pueden actuar directamente sobre los OC para promover su apoptosis33-35. La mayoría15,36,37, pero no todos los estudios38-40, detectan RE en OC. Además en otros tipos celulares relaccionados con el hueso también se han encontrado receptores esteroideos en células del estroma óseo (RA22,41, RE*22,42,43, REß22,43,44), en megacariocitos (RA, RE)37,41 y en células endoteliales del compartimento óseo (RA, RE)9,37,41.
Para aclarar el papel de los distintos tipos de receptores esteroideos en la homeostasis ósea se procedió al desarrollo de ratones transgénicos en los que se inactivaba de forma específica el gen de cada receptor y se estudiaba posteriormente su fenotipo óseo (ratones knockout) (KO). De este modo surgieron distintos tipos de roedores: ArKO (inactivación del gen de cyp19-aromatasa)45,46, BERKO (inactivación del gen de REß)47-49, ERKO (inactivación del gen de RE*)49,50, DERKO (inactivación del gen de ambos RE)51-55 y ANDRKO (inactivación del gen de RA)56,57. Aunque el significado exacto de los datos de estos ratones mutantes no está suficientemente esclarecido, existen varias hipótesis de interpretación.
Hueso trabecular. Era comúnmente aceptado que tanto los E como los A, tras ser aromatizados a E, actuaban sobre el hueso trabecular únicamente a través de RE. En ratones transgénicos hembra RE* desempeña un papel estimulador, mientras que REß es inhibidor, como lo demuestra el hecho de que las hembras BERKO posean mayor cantidad de hueso trabecular que el fenotipo de hembra silvestre. REß puede participar en la pérdida ósea relacionada con la edad en hembras, estimulando la resorción tanto endocortical como de hueso trabecular o inhibiendo el efecto estimulador de RE* en la formación ósea. De este modo, la deleción de REß puede aumentar la sensibilidad del hueso a RE* y, por lo tanto incrementar la acción de los E a pesar de la disminución de sus niveles séricos relacionada con la edad. En ratones macho la acción se produce a través de RE*, no teniendo ningún papel REß.
En los últimos años se ha descubierto también un modo de acción directo de los A a través del RA, al menos en roedores, tanto femeninos como masculinos, en los que ejerce una función protectora sobre el hueso trabecular.
Hueso cortical.Crecimiento longitudinal.En roedores en período de crecimiento tanto los A como los E parecen estimular el crecimiento longitudinal del hueso vía RE*. En ratones hembra REß ejerce un efecto inhibitorio, si bien en machos no parece mediar ninguna acción. De este modo, bajas concentraciones de E (como las que existen en varones) son estimuladoras, mientras que altas concentraciones (como en hembras) son inhibitorias del crecimiento longitudinal. El resultado de este proceso es una mayor longitud de los huesos en varones al final de la pubertad. El hecho de que los A únicamente actúen sobre el hueso trabecular a través de su aromatización a E lo demuestra el que el fenotipo óseo de los varones con deficiencia estrogénica (por mutación en el gen de la aromatasa) o resistencia estrogénica (por mutación del gen del RE*) se caracterice por ausencia de estirón puberal y retraso en el cierre epifisario.
En el momento actual permanece sin definir el papel del RA en este tipo de crecimiento y en el cierre epifisario tanto en humanos como en animales.
Crecimiento radial. Los A estimulan el crecimiento radial, mientras que los E disminuyen la aposición de hueso perióstico.
REß puede ser responsable, al menos en parte, del dimorfismo sexual esquelético, ya que las hembras BERKO poseen una mayor anchura de hueso cortical que el fenotipo de hembra silvestre. Estos cambios pueden ser el resultado del antagonismo mediado por REß a la formación de hueso perióstico estimulada por RE*. No se ha demostrado que REß tenga esta misma función limitante sobre el crecimiento radial en varones, ya que los ratones macho ERKO, DERKO, ArKO y ANDRKO experimentan una disminución del crecimiento radial, mientras que los ratones BERKO no.
En varones la acción de los andrógenos sobre el crecimiento radial se realizaría a través de RA y de RE*, si bien la importancia relativa de ambas vías y su interacción permanecen sin definir. Los datos disponibles son controvertidos, puesto que si bien la mayor parte de los estudios indican que RA es el mediador fundamental de la expansión perióstica (ratas Tmf [ratas macho feminizadas con testículos resistentes a los A] y ratones ANDRKO); otros sugieren que RE* también es un importante estimulador, ya que los inhibidores de la aromatasa en presencia de RA y niveles androgénicos normales detienen el crecimiento perióstico en ratas. Una posible explicación, todavía pendiente de ser confirmada, sería que al crecimiento del hueso perióstico contribuye fundamentalmente RA y con menor intensidad RE*. Esta hipótesis explicaría por qué la expansión perióstica puberal se detiene antes en la mujer que en el varón y por qué vuelve a reactivarse en mujeres posmenopáusicas.
MEDIADORES MOLECULARES DE LA ACCION DE LOS ESTEROIDES SEXUALES EN CÉLULAS OSEAS
Los primeros estudios apuntaban al papel del déficit estrogénico en el incremento de la producción de las citocinas proinflamatorias IL-1, IL-6, TNF*, factor estimulante de colonias de granulocitos-macrófagos (M-CSF) y prostaglandina-E2 (PGE2). Estas citocinas incrementan la resorción ósea, principalmente por aumentar el pool de preosteoclastos en médula ósea58. Los E también estimulan la producción de TGFß59, un inhibidor de la resorción ósea, cuyo principal reservorio en el organismo es el hueso, que actúa directamente en osteoclastos para disminuir su actividad y la tasa de apoptosis.
Sin embargo, la regulación estrogénica de la resorción ósea fue reevaluada tras el descubrimiento del sistema RANKL osteoprotegerina (OPG)60. La línea celular estroma-osteoblasto secreta OPG, que neutraliza el efecto de RANKL. Los E incrementan OPG61 y disminuyen M-CSF62 y RANK63. Parte de este efecto en el sistema de señalización puede ser indirecto por medio de intermediarios estimulados por E. Así, IL-1 y TNF* incrementan RANKL, OPG y M-CSF, mientras que PGE2 incrementa RANKL y disminuye OPG. Aún no se ha demostrado que los E regulen directamente RANKL o RANK. Por tanto, parece probable que los E inhiben la resorción ósea induciendo cambios pequeños pero acumulativos en múltiples factores E dependientes.
Se conoce menos acerca de los mecanismos moleculares de acción de los andrógenos sobre las células óseas. Existen tres factores reguladores de la acción androgénica expresados localmente en el hueso, dos de los cuales ya se han mencionado previamente al hablar de los E: TGFß, IGF e
IL-6.
Teóricamente los A preservan el hueso mediante la inducción de TGFß e IGF y la inhibición de IL-6. Varios estudios in vitro e in vivo han demostrado que los A
aumentan la expresión y/o la actividad de TGFß. De hecho, la orquectomía disminuye el contenido de TGFß del hueso64,65, mientras que el tratamiento con T lo aumenta. Se desconoce si este efecto tiene lugar a través de RE o RA. IGF1 e IGFBP estimulan la proliferación y diferenciación de OB. Los A regulan la expresión y/o la actividad de IGF directamente regulando IGF o indirectamente regulando IGFBP66. La DHT y la T suprimen la producción de IL-6 en cultivos de OB y de células estromales42,67. Más aún, los A inhiben la expresión de las subunidades gp80 y gp130 del receptor de la IL-668.
Otras vías paracrinas a través de las cuales los A pueden también regular el metabolismo óseo son la inhibición de la producción de PGE2 inducida por PTH e IL-269, el aumento de la producción de IL-1ß70, el aumento del efecto mitogénico del factor de crecimiento fibroblástico en cultivos de OB71 y efecto sobre la OPG72. Tanto en células osteoblasticas in vitro como en varones ancianos in vivo, los A disminuyen73 y los E incrementan61 la producción de OPG, lo que puede explicar en parte por qué la acción antirresortiva de la T es menor que la de los E. Quedan pendientes de investigar los efectos in vivo que tienen los esteroides sexuales sobre la OPG y su importancia fisiológica en la regulación de la homeostasis ósea.
Finalmente, Kousteni et al74 han demostrado que los efectos antiapoptóticos de E y T en osteoblastos y osteocitos pueden ser mediados por señalización rápida, no genómica y no dependiente del sexo a través del dominio de unión a ligando de RE*, REß o RA. Esta acción, como veremos más adelante es distinta de las acciones clásicas de estos receptores, las cuales son dependientes del sexo, genómicas y transcripcionales.
Interacción con fuerzas biomecánicas
Frost75 ha insistido en el papel que las tensiones biomecánicas, especialmente las inducidas por contracción muscular, desempeñan en el nivel de masa ósea. Las tensiones son detectadas por un mecanostato interno del esqueleto que inicia cambios en el remodelado óseo para ajustar la masa ósea y su distribución a un nivel adecuado al ambiente de fuerzas biomecánicas. Bajos niveles de tensión mantenidos crónicamente conducirán a un modelo de «desuso» en el remodelado óseo. En dicho modelo se halla incrementado el turnover en todas las superficies óseas, pero en las superficies endosteales, aquellas en contacto con la médula ósea, se reabsorbe más hueso del que se forma. Este disbalance en el remodelado de la superficie endosteal inducido por la inactividad es mediado por un factor liberado por la médula ósea, llamado Rho. Los cambios histomorfométricos inducidos tanto en el déficit de E como en el modelo de «desuso» son muy similares. En ambos, la pérdida de hueso se halla confinada a la superficie endosteal y no implica a las superficies intracortical y periosteal76. Frost formuló la hipótesis de que el déficit de E altera el funcionamiento del mecanostato, disminuyendo la sensibilidad a las señales de tensión.
Las teorías de Frost se han visto confirmadas por varios estudios experimentales. Existe la evidencia de que «el mecanostato» son, al menos en parte, los osteocitos, que contienen Res77. Rho comprende una cascada de citocinas derivadas de la médula ósea que incluye las citocinas proinflamatorias y el sistema regulador OPG/RANKL/RANK. Estas citocinas regulan la diferenciación de osteoclastos y osteoblastos desde precursores celulares61, y dichos precursores celulares en la médula ósea responden a E. Por tanto, es plausible que las citocinas de la médula ósea medien los efectos de los E y de las tensiones biomecánicas. También se ha demostrado experimentalmente la interacción entre las fuerzas mecánicas y los E, puesto que el efecto de las tensiones mecánicas puede ser bloqueado por tratamiento con los antagonistas de RE ICI 182,780 o tamoxifeno78.
Recientes estudios in vitro sugieren que los esteroides sexuales y las cargas mecánicas ejercen sus efectos a través de vías similares. De hecho, se ha demostrado que RE* media en ambos sexos la proliferación de los osteoblastos inducida por la carga mecánica79,80. La importancia de esta interacción entre E sexuales y carga mecánica in vivo no se encuentra suficientemente esclarecida en la actualidad.
Efectos no genómicos y no dependientes del sexo de los esteroides sexuales
Las hormonas sexuales también actúan por medio de efectos no genómicos, que implican acciones a nivel de la membrana celular o del citoplasma81. Kousteni et al82 demostraron que E y A actúan de una forma rápida, no genómica para activar la vía ERK1/2 que aumenta la supervivencia de OB en presencia de agentes apoptóticos como el etopósido. De este modo se comprobó que tanto 17ß-E2 como 5*-DHT eran igualmente eficaces bloqueando el proceso apoptótico y que ambos podían ejercer su acción a través de RA y RE indistintamente. Estos hallazgos sugieren que un mecanismo sexo-inespecífico en ambos receptores puede ser activado por E o A con consecuencias aparentemente idénticas. Usando los E como paradigma, los autores muestran que la actividad antiapoptótica de RE es preservada cuando el receptor se halla en el citoplasma o en la membrana de la célula, pero se pierde si se halla en el núcleo. Mientras la acción antiapoptótica de RE no requiere la unión al ADN, su acción sobre la regulación de la transcripción precisa de la activación del dominio de unión a ligando (LBD) del RE para unirse al ADN. Finalmente, los autores demuestran que los efectos antiapoptóticos de los E pueden disociarse de sus efectos transcripcionales empleando el nuevo ligando sintético del RE llamado estren, que modula sólo su actividad antiapoptótica.
Estos estudios sugieren un nuevo mecanismo de acción similar para E y A en la biología de los OB, tanto en varones como en mujeres, y son prometedores, pero surgen varias preguntas de naturaleza técnica y biológica al respecto. Desde el punto de vista biológico: ¿cuál es el significado del papel de E y A sobre la supervivencia de los OB in vivo?, ¿son las acciones biológicas de E y A aquí mencionadas representativas de lo que sucede in vivo? Desde el punto de vista técnico surgen otras cuestiones. Los hallazgos del estudio sugieren que E2 y 5*-DHT suprimen la apoptosis en OB y que dicha supresión es revertida de modo no específico tanto por el antagonista del RE ICI 182,780 como por el antagonista del RA flutamida (F). Estos hallazgos son sorprendentes en vista de la fuerte selectividad por el receptor característica tanto de ICI como de F. Otro hallazgo muestra que los efectos antiapoptóticos, tanto de E2 como de 5*-DHT en células HeLa pueden ser mediados tanto por RE como por RA. Estos descubrimientos desafían nuestros conceptos tradicionales acerca de la selectividad, especificidad y acción de los esteroides sexuales y nos inducen a buscar nuevos mecanismos de acción.
¿Cuáles son las señales intermediarias disparadas por E y A para iniciar la respuesta antiapoptótica en osteoblastos? El grupo de Manolagas ha hallado, tanto in vitro como in vivo, datos que sugieren que el efecto no genómico de las hormonas sexuales implica una regulación mediada por cinasas de los factores de transcripción comunes. Así, los efectos no genómicos de los esteroides sexuales influyen en la actividad de Elk-1, la proteína-ß que se une al enhancer CCAAT (C/EBPß), y proteínas reguladas por cAMP, o c-Jun/c-Fos por una acción extranuclear del RE o RA, llevando todo ello a la activación de la vía Src/Shc/ERK o a la regulación a la baja de la cinasa N-terminal de C-Jun respectivamente. Un hallazgo interesante es que el ligando sintético estren, que reproduce los efectos no genómicos de las hormonas sexuales sin afectar a la transcripción clásica, incrementa la densidad mineral ósea (DMO) en ratones hembra ovariectomizadas y en ratones macho orquidectomizados, sin afectar a los órganos reproductivos (útero y vesículas seminales). Tales ligandos se deben investigar como potenciales alternativas terapéuticas en la sustitución hormonal para el tratamiento de la osteoporosis, tanto en mujeres como en varones.
FUNCION DE LAS HORMONAS SEXUALES EN EL CRECIMIENTO Y LA MADURACION DEL ESQUELETO
Antes de la pubertad niveles basales de GH/IGF-I mantienen un lento, pero continuo, crecimiento óseo. La pubertad es desencadenada por un incremento en la secreción hipotalámica de GnRH, lo que estimula la síntesis de gonadotropinas y, por tanto, incrementa la secreción gonadal de hormonas sexuales83. A su vez, los E y A estimulan la secreción pulsátil de GH y secundariamente de IGF-I en ambos sexos actuando a través de RE*, como lo demuestran las bajas concentraciones séricas de IGF-I existentes en ratones ERKO de ambos sexos. Los A ejercen este papel tras ser aromatizados a E a nivel central. De hecho, los inhibidores de la aromatasa84 causan una disminución de los niveles de IGF1, mientras que el antiestrógeno ICI 182,780, que no atraviesa la barrera hematoencefálica, no modifica sus concentraciones en ratas masculinas85.
El incremento de GH, IGF-I y E actúa de modo coordinado para proporcionar apoyo al brote de crecimiento puberal. Sin embargo, es la elevación de los niveles séricos de E la responsable última de este crecimiento. Los varones con mutaciones homocigotas en RE* o en genes de la aromatasa no realizan un rápido crecimiento durante la adolescencia a pesar de niveles normales o incrementados de T86. Por otra parte, el incremento continuo de los niveles de E durante la pubertad es la causa del cierre epifisario en ambos sexos. Como los niveles estrogénicos son más altos en las mujeres, el cierre epifisario ocurre antes en ellas, por lo que los varones alcanzarán una mayor longitud ósea. Por tanto, los estrógenos por una parte inician la fase de crecimiento puberal, y por otra la finalizan por medio de la inducción del cierre epifisario. Esto unido a la acción de los esteroides sexuales sobre el crecimiento radial del hueso, comentada anteriormente, genera después de la pubertad un dimorfismo sexual esquelético que conlleva una mayor longitud ósea, perímetro externo, perímetro interno y volumen cortical en varones que en mujeres.
Las hormonas sexuales también parecen incrementar la masa ósea durante la maduración del esqueleto independientemente de los efectos ejercidos sobre el eje GH-IGF-I. La masa ósea es un 25% mayor en varones pospúberes que en mujeres pos-púberes, en relacción probablemente con los niveles de T sérica, ya que el incremento en los niveles de GH e IGF-I es similar o incluso mayor en chicas que en chicos. Así, los cambios inducidos por los A en el crecimiento y la composición corporal también pueden tener un gran impacto sobre el esqueleto. En ratas macho orquectomizadas se detecta una disminución de la masa muscular y un aumento de la masa grasa87,88. Esta pérdida muscular podría causar una reducción del estrés mecánico sobre el hueso. Por tanto, la disminución del crecimiento radial y longitudinal que acontece tras practicar una gonadectomía podría suponer una adaptación del hueso en crecimiento a los cambios inducidos por la orquectomía en las fuerzas mecánicas, más que en la acción directa de los esteroides sexuales sobre el hueso. Al contrario, el aumento del crecimiento esquelético en respuesta a los A podría reflejar un aumento de la carga mecánica.
En línea con las afirmaciones anteriores se han encontrado varios puntos de unión entre los adipocitos y el hueso, existiendo evidencias de que algunas adipocitocinas (como la leptina) son las mediadoras de esta unión vía sistema nervioso central (fundamentalmente a través del núcleo paraventricular)89.
La pubertad en varones con respecto a mujeres se caracteriza no sólo por un mayor crecimiento longitudinal, sino también por una mayor ganancia de masa muscular90. El aumento de masa muscular incrementa las fuerzas biomecánicas que actúan sobre el hueso91. Es por tanto tentador especular, basándonos en la teoría de Frost anteriormente expuesta, que los cambios en la composición corporal (aumento de masa muscular, disminución de grasa) contribuirían a conseguir un mayor tamaño óseo en varones. De hecho, en varones con hipogonadismo (HG) el tratamiento con T provoca cambios en la composición corporal, pero las implicaciones esqueléticas que esto pueda tener todavía deben ser estudiadas92.
EFECTOS DE LOS ANDROGENOS EN EL ESQUELETO HUMANO
En 1940 Albright y Reifenstein fueron los primeros en hacer referencia a sus propiedades antiosteoporóticas y anabólicas93, si bien la mayor parte de los esfuerzos de investigación realizados desde entonces se han enfocado fundamentalmente hacia el campo de los E, dado el gran problema de salud pública que supone la osteoporosis en la mujer.
EFECTOS DE LOS ANDROGENOS EN EL ESQUELETO FEMENINO
El papel de los A en la homeostasis del esqueleto femenino no está bien establecido.
En las mujeres las concentraciones de A varían considerablemente; si bien la concentración de T es siempre menor que en varones, las concentraciones de otros A más débiles como la dehidroepiandrosteronasulfato (DHEA-S) y la androstenediona son iguales94. Los A, de este modo, podrían contribuir a las diferencias clínicamente significativas que existen en la DMO entre las distintas mujeres.
Los A podrían estimular el desarrollo esquelético durante la pubertad, según se deriva del hallazgo de una mayor DMO en mujeres con síndrome de ovario poliquístico (PCOS). Esto ha sido confirmado mediante pQCT, reflejando, por tanto, cambios auténticos en la composición del tejido óseo y no sólamente cambios en el tamaño del hueso. Se desconoce todavía si esta acción estimuladora de los A ocurre a través del RA o a través de su aromatización a E. Las mujeres obesas con PCOS tienen mayor DMO que las delgadas, lo cual sugiere que la aromatización de los A en el tejido adiposo desempeña un papel importante. Permanece sin esclarecer la importancia que otras alteraciones metabólicas presentes en PCOS (disminución de niveles de SHBG, hiperinsulinismo) podrían tener en la homeostasis ósea de estas mujeres. Por último, las diferencias en la composición corporal podrían ser determinantes clínicamente importantes de la integridad esquelética, como lo sugieren el hallazgo de diferencias en la DMO dependientes de la localización esquelética en estas mujeres95.
Un punto de especial interés consiste en el estudio de los efectos que sobre el hueso ejercen los distintos tratamientos empleados en el hirsutismo, particularmente los agonistas de GnRH y los antagonistas de RA, solos o en combinación. El antagonista de RA flutamida no induce cambios en la densidad de la columna vertebral lumbar96, mientras que sí disminuye la DMO cuando se administran conjuntamente espironolactona y linesterol97. La supresión de los niveles de esteroides sexuales usando análogos de GnRH induce una disminución de la densidad ósea semejante a la que acontece en mujeres posmenopaúsicas98. Sorprendentemente, la espironolactona (pero no la flutamida) mantiene la DMO en mujeres hirsutas en tratamiento con análogos de GnRH, como se demostró en un estudio de 6 meses de duración99. El mecanismo por el que se produce este efecto protector es desconocido y está en contraposición con el resto de estudios en los que la misma asociación ocasiona disminución de DMO.
En mujeres posmenopáusicas está menos estudiado el efecto de los A sobre el hueso. En la menopausia se produce un descenso del 70% en el nivel de A séricos (especialmente DHEA-S)94,100, pero la importancia que esto pueda tener en la pérdida de masa ósea y en el riesgo de fracturas no está demostrada mediante estudios prospectivos. En estudios transversales no se ha encontrado una relación consistente entre los niveles séricos de DHEA-S y la DMO101,102. En la actualidad no está claro si el modo de acción de estos A adrenales sobre el esqueleto femenino es a través del RA o de su aromatización a E. Por último, haremos una breve referencia al papel potencial que la terapia sustitutiva con T puede tener en estas mujeres103,104. La combinación de dosis bajas de T y E da lugar a un aumento adicional de DMO si se compara con la administración aislada de E104,105. Sin embargo, mientras no se compruebe la seguridad de este tratamiento combinado a largo plazo, no puede ser recomendado de forma sistemática.
EFECTOS DE LOS ANDROGENOS EN EL ESQUELETO MASCULINO
En esta sección se revisarán los efectos esqueléticos de la castración, el HP y la resistencia androgénica.
La castración quirúrgica y química (agonistas de GnRH) induce una disminución de los niveles de esteroides sexuales en el varón, que se sigue de una rápida pérdida de hueso. Se produce una disminución de la DMO en columna vertebral lumbar de un 5-10% en el primer año tras la castración, continuando posteriormente la pérdida ósea de forma lineal. Recientemente se ha comprobado que también ocurre una disminución de DMO clínicamente significativa, aunque de menor intensidad, en cadera y radio. Además, la deficiencia de A produce cambios en la composición corporal, que también pueden contribuir de forma independiente a un aumento en el riesgo de fracturas. Todo esto sugiere que el mecanismo de pérdida ósea en varones con deficiencia androgénica es similar al que ocurre en situaciones de insuficiencia gonadal en mujeres y animales, es decir, se produce un disbalance a favor de la resorción ósea, lo que ocasiona una pérdida de hueso, especialmente trabecular. Falta todavía por confirmar histomorfométricamente el aumento en el turnover óseo de estos varones, pero se ha comprobado que la supresión del turnover preserva la DMO (los bifosfonatos previenen la pérdida ósea en pacientes con cáncer de próstata sometidos a castración química).
El HG se define como una concentración de T baja de forma mantenida. Los varones con HG presentan una DMO menor que los varones normales (particularmente en áreas de hueso trabecular, por ejemplo, la columna vertebral). Algunos estudios sugieren que la resorción ósea, y en menor medida la formación, están aumentadas, si bien otros trabajos aportan datos histomorfométricos de baja formación ósea106,107. Se ha detectado mediante resonancia magnética nuclerar (RMN) un deterioro de la arquitectura del hueso trabecular de la porción distal de la tibia en estos varones108. Varios experimentos demuestran un metabolismo del calcio y de la 1,25(OH)2 VitD3 normal, lo cual sugiere que el HG puede afectar la homeostasis ósea, independientemente del calcio y de la vitamina D. Experimentos realizados con el modelo de rata orquidectomizada apoyan esta teoría. Si bien existen estudios casos/controles que demuestran una prevalencia mayor de la esperada de HG en varones con fractura de columna vertebral o de cadera109,110, faltan estudios prospectivos que establezcan la existencia de una relación causa-efecto entre ambas variables.
El HG masculino puede deberse a una gran variedad de enfermedades, que a su vez pueden tener cierto efecto deletéreo sobre el esqueleto. No obstante, diferentes estudios han hallado la misma disminución de DMO en todas las etiologías causantes de HG, sugierendo la posibilidad de que el HG per se y no la enfermedad primaria que lo origina sea la responsable de la pérdida ósea111. En todas las formas de HG estudiadas (pubertad retrasada, HG hipogonadotropo, HG por hiperprolactinemia, Sd. de Kilnefelter [SK]) se comprueba una disminución de DMO en columna vertebral, radio y fémur. En pacientes con HG hipogonadotropo el análisis del turnover óseo ha producido resultados contradictorios, con datos histomorfométricos de osteoporosis de bajo recambio en algunos pacientes112, pero niveles elevados de marcadores de formación y resorción ósea en otros113. En el HG por hiperprolactinemia la corrección del HG aumenta de forma significativa la densidad cortical ósea, independientemente de los niveles de prolactina, sugiriendo que es la deficiencia androgénica y no el exceso de prolactina lo que altera la homeostasis ósea en estos pacientes114.
Recientemente, el papel de los E en el desarrollo y mantenimiento del esqueleto en ambos sexos ha recibido especial atención. El HG debe considerarse como una combinación de varios grados de deficiencia androgénica y estrogénica, que dañarían el hueso de forma diferente. Más aún, el grado de deficiencia estrogénica en el HG masculino puede estar en relación con la capacidad de aromatización de los A, de manera que los pacientes con niveles androgénicos muy bajos tienen además una capacidad limitada para su aromatización.
Los varones con síndrome de insensibilidad completa a los A (cASI) tienen menor DMO en columna vertebral y cadera si se comparan con controles. Estos hallazgos sugieren que los A pueden actuar directamente a través del RA. Marcus et al115 afirman que el nivel de E es un importante determinante de la DMO en estos pacientes, ya que se ha comprobado que aumentan la DMO en columna vertebral y cadera116,117. Finalmente, la resistencia androgénica en humanos no se ha asociado con alteraciones en el crecimiento longitudinal, lo que llama la atención sobre la importancia de los E en este tipo de crecimiento y en el cierre epifisario en varones.
CONCLUSIONES
Se están comenzando a investigar las bases autocrinas y paracrinas de la acción de los esteroides sexuales sobre el hueso, siendo necesarios nuevos estudios para definir la implicación de diversas citocinas en este proceso.
En cuanto al efecto que sobre el hueso tiene la interacción de los esteroides sexuales con las fuerzas biomecánicas, están surgiendo nuevas hipótesis acerca de que los cambios inducidos en la composición corporal por los esteroides sexuales serían en parte responsables del dimorfismo sexual esquelético tras la pubertad.
Recientemente se ha demostrado la existencia de RA en casi todas las células óseas y en el cartílago de crecimiento, lo que abre la posibilidad de que los A puedan actuar sobre el hueso no sólo a través de su aromatización a E y su interacción con RE, sino de forma directa utilizando su propio receptor. La activación de RA estimula el crecimiento del hueso trabecular y el crecimiento radial del hueso cortical. Por otra parte, RE* favorece el crecimiento tanto cortical como trabecular, mientras que REß ejerce un papel inhibidor en mujeres, no desempeñando aparentemente ninguna función en varones, lo que podría explicar en parte el dimorfismo sexual del esqueleto adulto.
En vista del papel esencial del RE*, algunos SERM (moduladores selectivos de RE*) con la apropiada selectividad de tejido podrían ser útiles en el tratamiento de varones ancianos con osteoporosis; y viceversa, moduladores selectivos de RA podrían ser igualmente interesantes en el campo de la enfermedad metabólica ósea femenina. Nuevas tecnologías genéticas y proteómicas combinadas con la disponibilidad de un completo set de ratones transgénicos knockout podrían proporcionar nuevas informaciones al respecto a corto plazo.
El descubrimiento de que tanto E2 como 5*-DHT suprimen la apoptosis de OB y que dicha supresión es revertida de modo no específico tanto por el antagonista del RE, ICI, como por el antagonista del RA, flutamida, desafían nuestros conceptos tradicionales acerca de la selectividad y especificidad de la acción de los esteroides sexuales sobre el hueso. En la misma línea, Kousteni et al proporcionan nuevos e interesantes datos que demuestran que las hormonas sexuales protegen a los OB por medio de un nuevo mecanismo de naturaleza no genómica y no dependiente del sexo. Por tanto, respondiendo a la pregunta que plantea el título de esta revisión podemos concluir que, a la vista de los datos disponibles en el momento actual, el efecto de los esteroides sexuales sobre el hueso es independiente del sexo. El hallazgo de un mecanismo sexo-inespecífico para el funcionamiento de RA y RE por el que éstos pueden ser activados tanto por E como por A con consecuencias aparentemente idénticas abre una importante vía de investigación en el futuro con implicaciones terapéuticas sumamente interesantes.