





INTRODUCCIÓN
La osteopetrosis es un heterogéneo grupo de entidades hereditarias que consisten en el aumento de la masa y densidad ósea («hueso dentro de hueso») definido ya en 1904 en la descripción del radiólogo alemán Albers-Schönberg1.
La patogenia es un fallo de la reabsorción ósea y desequilibrio entre la actividad osteoclástica y la osteoblástica que desencadena una alteración de la remodelación, lo que genera una menor actividad del factor estimulante de colonias macrofágicas (MCSF), interleucina 1 e interleucina 6, que en condiciones fisiológicas son necesarios para la diferenciación de células precursoras de osteoclastos2; todo sobre una base de mutaciones génicas CLCN, CAII y TCIRG1.
DESCRIPCIÓN DE LOS CASOS
En nuestro hospital entre 1985 y 2005 se han diagnosticado cuatro casos de osteopetrosis en adulto de los cuales uno ha fallecido de insuficiencia cardíaca por cardiopatía isquémica crónica y tres están en seguimiento durante 28, 4 y 22 años, respectivamente. La edad del diagnóstico oscila entre los 50 y 84 años, y un caso se diagnosticó a los 5 años (actualmente tiene 33) con afectación de calota, huesos largos, vértebras y pelvis, sin insuficiencia de médula ósea, ni osteomielitis, ni fracturas, ni afectación de pares craneales, encontrándose asintomático en la actualidad. Son tres mujeres y un varón. Han tenido fracturas tres de los cuatro pacientes (dos de huesos largos y uno vertebral). La osteomielitis sólo se ha dado en un paciente. La forma de presentación en un caso fue hallazgo casual, otro manifestaba dolor óseo y en los otros dos las fracturas por caída fueron las que condujeron al diagnóstico. En cuanto a las alteraciones analíticas la fosfatasa alcalina se eleva en tres pacientes, la hormona paratiroidea (PTH) sólo en uno y la calcemia y fosfatasa ácida permanecieron normales en los cuatro (tabla 1).
DISCUSIÓN
Hay formas que se heredan con carácter autosómico recesivo, se asocian con trastornos hematológicos como fallo medular, anemia y hepatoesplenomegalia por hematopoyesis extramedular, también con trastornos neurológicos como hidrocefalia y afectación de pares craneales con atrofia óptica, alteraciones en la motilidad ocular, sordera y parálisis facial, siendo menos frecuentes la anosmia y las lesiones del trigémino. Son las formas infantiles las que conducen a la muerte antes de que se cumpla la primera década de la vida (osteopetrosis infantil maligna). En el ser humano el locus geni parece que se ha identificado en el 11q13.
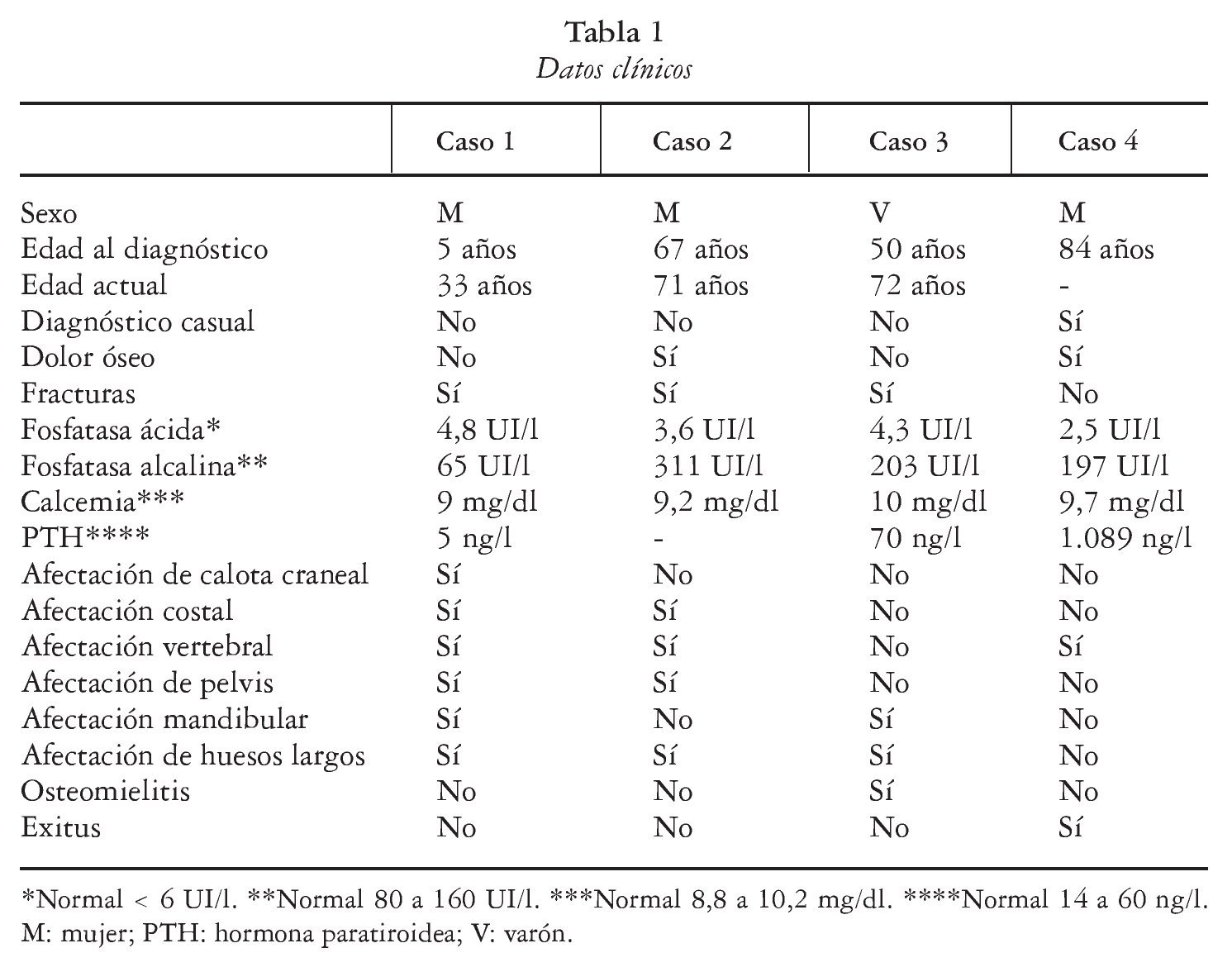
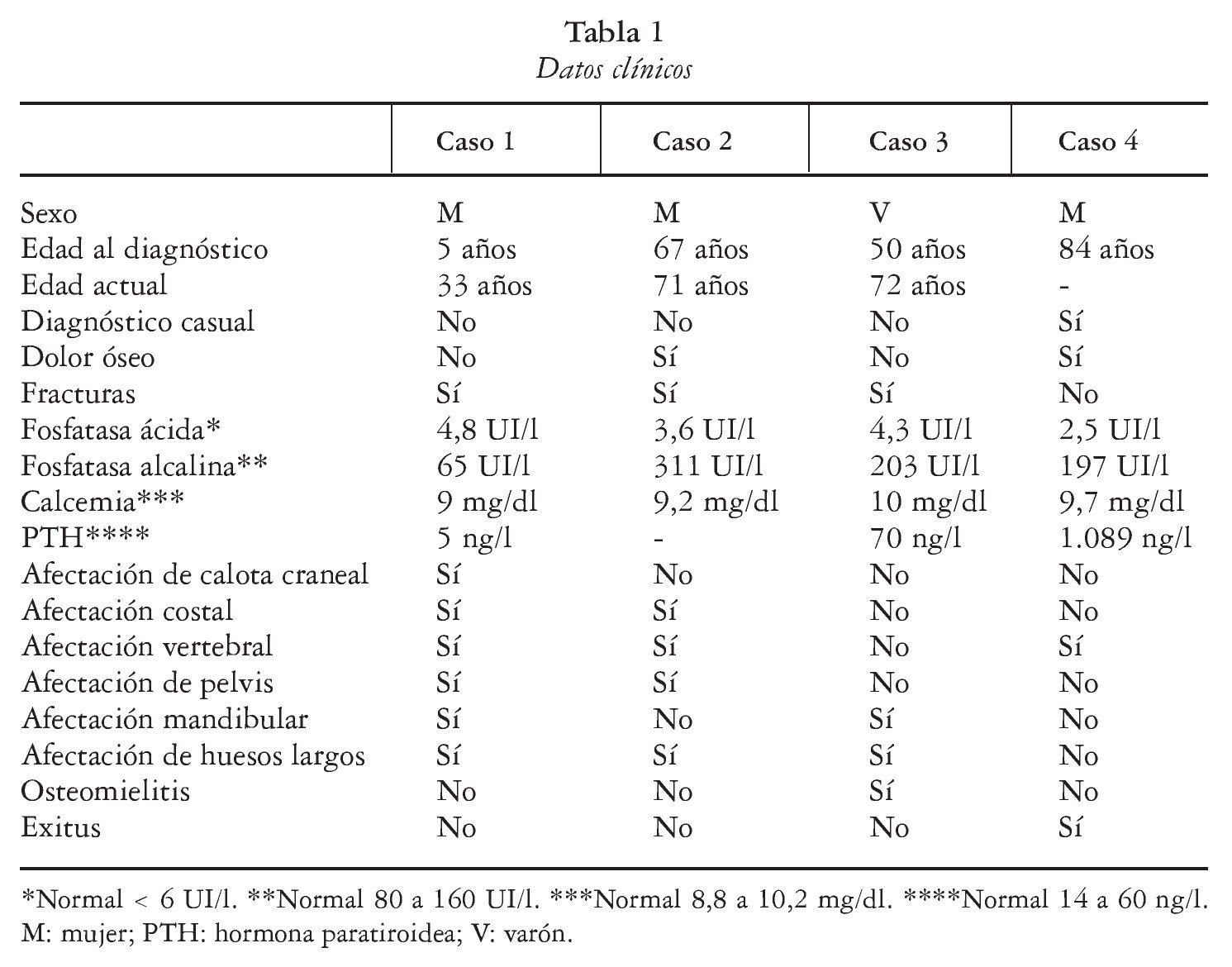
Se conocen cada vez mejor las interacciones entre las células osteoblásticas/estromales y las células hematopoyéticas precursoras de los osteoblastos que dan lugar a la formación de osteoclastos. Un miembro de la superfamilia de los receptores del factor de necrosis tumoral (TNF), llamado osteoprotegerina (OPG), actúa como señuelo que se une, y presumiblemente neutraliza, al ligando RANK (RANKL). Este complejo se une a RANK (receptor transmembranoso de las células progenitoras de los osteoclastos) activando su diferenciación y su función (fig. 1). Los ratones deficientes en RANK carecen de osteoclastos y presentan una intensa hiperdensidad ósea2.
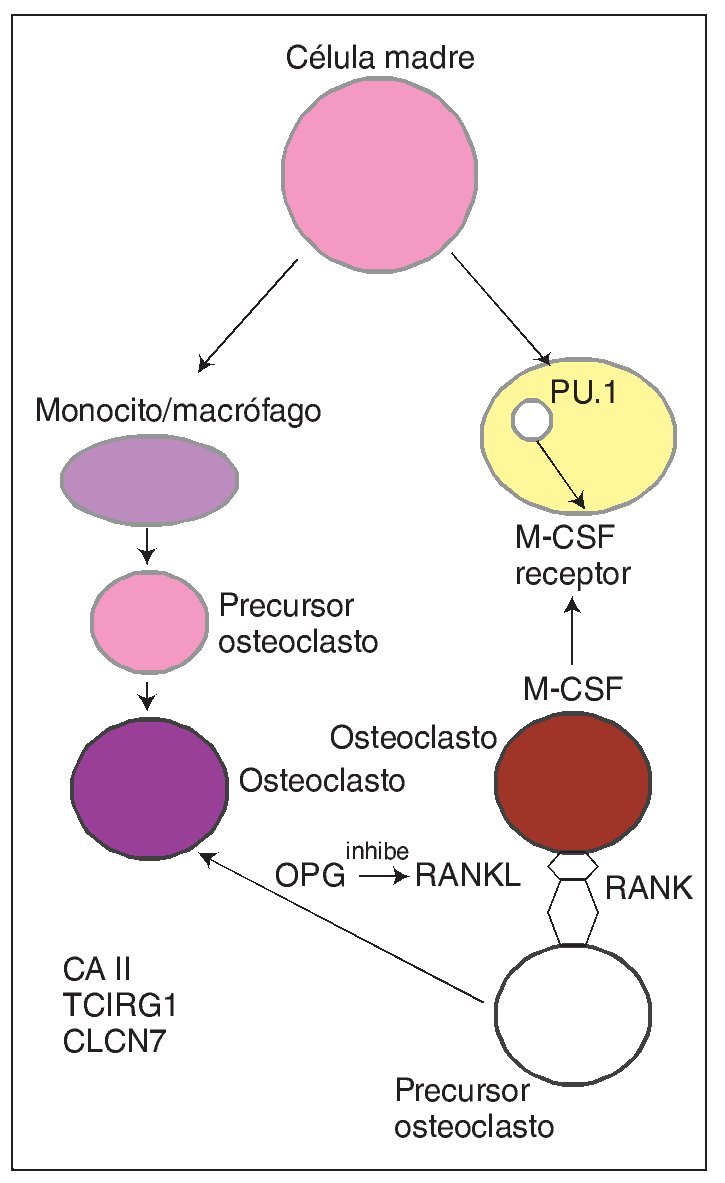
Fig. 1. En experimentación animal (ratones) la osteopetrosis se ha asociado a defectos en la citoquina M-CSF (factor estimulante de colonias macrofágicas) responsable de la diferenciación de precursores osteoclásticos y del factor de transcripción de esta citoquina, PU.1, así como alteraciones en la interacción RANK (receptor activador del factor nuclear κB) y RANKL (ligando del RANK) y otros mediadores implicados. En la especie humana la enfermedad se relaciona con mutaciones génicas que afectan a CA II, canales de cloro (gen CLCN7) y bomba de protones (gen TCIRG1) y más recientemente también por mutación del gen RANKL. OPG: osteoprotegerina.
Con carácter autosómico recesivo se hereda también una forma de osteopetrosis ligada a una disfunción de la enzima anhidrasa carbónica II (fig. 2), que es el componente esencial del sistema enzimático que interviene dentro del metabolismo del osteoclasto en la formación de ácido carbónico y de hidrogeniones que facilitan la acidificación del medio y la descomposición de los minerales óseos como la hidroxiapatita. Pues bien, una baja actividad de la misma hace que los osteoclastos no ejerzan su función de reabsorción ósea y, por tanto, que aumenten la masa y la densidad del esqueleto. Se asocia a acidosis tubular renal, baja estatura, compresión de pares craneales, trastornos de desarrollo de la dentición, fracturas frecuentes y a veces osteomielitis. Es la forma intermedia de osteopetrosis que se ha descrito en varias familias, que se da en la adolescencia y es menos grave que la forma infantil ya que no suele asociar insuficiencia medular.
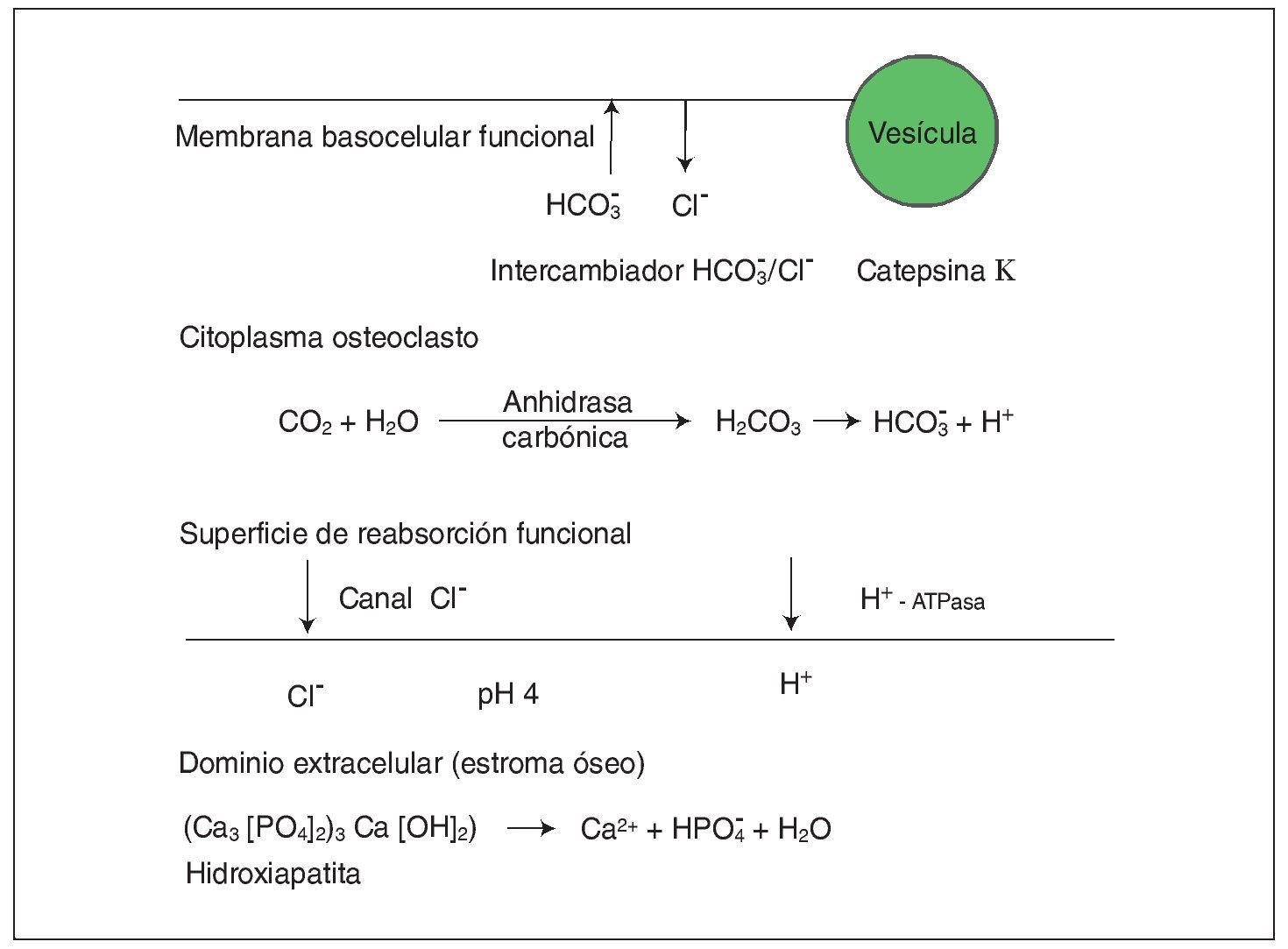
Fig. 2. En el citoplasma del osteoclasto se produce fisiológicamente la transformación mediada por la enzima anhidrasa carbónica II de CO2 y H2O en CO3H2, que posteriormente se disocia en bicarbonato y protones. Los protones que atraviesan la membrana rugosa son transportados a la superficie de reabsorción dando lugar a un dominio extracelular óseo ácido propicio para la destrucción del hueso (degradación de hidroxiapatita). Paralelamente la neutralidad de la membrana rugosa está preservada por un canal de cloro. En el otro dominio funcional (membrana basocelular) se elimina el exceso de CO3H- intercambiando con Cl-; se produce la exocitosis de productos de desecho mediada por vesículas y actúa la catepsina K degradando matriz orgánica.
Smahi y Döffinger describieron en 2002 dos casos infantiles de osteopetrosis ligada al cromosoma X3 asociada a displasia ectodérmica, linfoedema inmunodeficiencia (síndrome DL-EDA-ID) por una mutación hipomórfica del NEMO (modulador esencial del NF-κB).
La osteopetrosis del adulto se hereda con carácter autosómico dominante y se distinguen dos tipos:
1. Tipo I: afecta a la calota craneal.
2. Tipo II: afecta a la base de cráneo, vértebras, pelvis y huesos largos.
Ambas son asintomáticas en la mayoría de los casos y de diagnóstico habitualmente casual; si bien hay veces, fundamentalmente en el tipo II, que el dolor óseo o una fractura adelantan el diagnóstico y otras veces una osteomielitis o la compresión de pares craneales, como en la forma recesiva, la manifiestan. En estos casos la mutación génica implicada es la misma que en la forma recesiva (CLCN7) (tabla 2).

En relación con lo que ya hemos referido anteriormente en la patogenia, también se ha descrito en humanos osteopetrosis debido a un defecto en el gen que codifica RANKL, caracterizado por un descenso en el número de osteoclastos y una progresión clínica más lenta. Se ha sugerido que estos pacientes podrían beneficiarse de tratamiento con RANKL recombinante4.
Para finalizar, los últimos estudios publicados sugieren la retirada de la clasificación clásica de la osteopetrosis del adulto tipo I, debido al fundamento etiopatogénico de que no es un defecto osteoclástico su causa sino una mutación del gen RLP55 que produce un defecto intrínseco del osteoblasto, y consideran que se reclasifique como una enfermedad de «masa ósea aumentada»6.
Correspondencia: F.J. Cabrera Aguilar. Hospital General Gregorio Marañón.
C/ Doctor Esquerdo, 46.
28007 Madrid. España.
Correo electrónico: fcabrera.hgugm@salud.madrid.org