El mantenimiento de la concentración de calcio iónico (Cai) extracelular en un nivel fisiológico virtualmente constante alrededor de 1,0 mM es una de las principales prioridades para los organismos terrestres por su papel clave en diversos procesos celulares como el control de la secreción exocrina y endocrina, la proliferación y la diferenciación celular y el mantenimiento del potencial de membrana. La concentración de Cai intracelular en reposo es 10.000 veces inferior (100 nM) pero aumenta hasta 1 mcM por la activación celular con liberación desde depósitos intracelulares y por entrada desde el exterior de la célula. Estos sistemas de transporte de calcio dan forma a las señales producidas por hormonas y neurotransmisores. Hoy es evidente que las señales de calcio son un código o lenguaje intracelular heterogéneo que se adapta a las funciones de cada tipo celular1.
El sistema hormonal que mantiene esa constancia requiere la actuación integrada de las hormonas calciotropas entre las que, en mamíferos, las glándulas paratiroideas tienen un papel central. En el hiperparatiroidismo primario (HPP) se producen anomalías en la regulación de la secreción de parathormona (PTH) y de la proliferación de las células paratiroi deas, describiéndose un aumento del número de células parenquimatosas y de células activas, así como un aumento del set-point de secreción de PTH mediado por Cai extracelular. Se han detectado diversas mutaciones, tanto de células germinales como somáticas, asociadas a HPP2. Se trata de mutaciones inactivadoras de genes de supresión tumoral (gen MEN-1, p53 y del retinoblastoma) o de mutaciones activadoras de protooncogenes (ciclina D1, gen RET). En los últimos años se ha querido implicar la presencia de mutaciones inactivadoras del receptor sensible a calcio (RSCa) en la génesis del HPP no familiar.
La clonación de fragmentos de ADN bovino funcionalmente activo en los oocitos de Xenopus laevis permitió aislar el RSCa3, que presenta una elevada homología entre diversas especies y tejidos (90%). Su descubrimiento probó que el Cai extracelular puede, de hecho, ser un primer mensajero extracelular como PTH, vitamina D u otras hormonas calciotropas. Está presente en la mayoría de otros tejidos relacionados con la homeostasis del calcio además de en otros no relacionados. El RSCa consta de tres dominios topológicos: extracelular (612 aa), hidrofóbico (250 aa con siete dominios transmembrana) y C-terminal intracelular (217 aa) en el que se han descrito polimorfismos. El dominio N-terminal está glucosilado con manosa o carbohidratos complejos (aunque la glucosilación no es imprescindible para su función). Pertenece a clase C, grupo II de la superfamilia de los receptores acoplados a proteínas G, presentando baja homología con los receptores del grupo I y III. Parece representar, evolutivamente hablando, una proteína de fusión entre el dominio extracelular N-terminal derivado de la antigua familia de las proteínas de unión a solutos (similar a las proteínas de unión periplásmica bacterianas) y la zona de la serpentina de membrana acoplada a proteínas G, con el fin de transducir las señales extracelulares al interior de las células eucariotas4,5.
El principal ligando del RSCa para reducir la secreción de PTH es el Cai extracelular. El magnesio también se liga al receptor aunque con dos o tres veces menor afinidad (probablemente se trate de un agonista fisiológico para modular la reabsorción renal de cationes divalentes). También se ligan al RSCa el gadolinium y policationes orgánicos (neomicina, espermina, espermidina) que están en altas concentraciones en algunas zonas del sistema nervioso donde pueden modular el microambiente. Además de estas funciones el RSCa es un sensor de carga o potencia iónica, por lo que integra una gran variedad de señales del microambiente inmediato que permite la elaboración de respuestas biológicas relevantes en las células que lo expresan5.
El RSCa se expresa en paratiroides, células C del tiroides, riñón, hueso e intestino. El mismo estímulo que inhibe la secreción de PTH, activa la de calcitonina (CT) y a la inversa. En el riñón parece auto-rregular la reabsorción de los cationes divalentes. En el hueso regula el recambio óseo esquelético pasando en microzonas concretas de resorción a formación ósea, mientras que en el intestino puede regular la motilidad y la absorción de calcio. El RSCa, por lo tanto, no sólo es mediador del clásico feed-back Calcio/PTH/CT sino que además participa en el feed-back de asa corta por el que el Cai reabsorbido en riñón, absorbido en intestino o resorbido en el hueso puede regular localmente esos mismos procesos y otros implicados en la homeostasis del calcio. Así pues, el Cai se debe considerar tanto un primer como un segundo mensajero5.
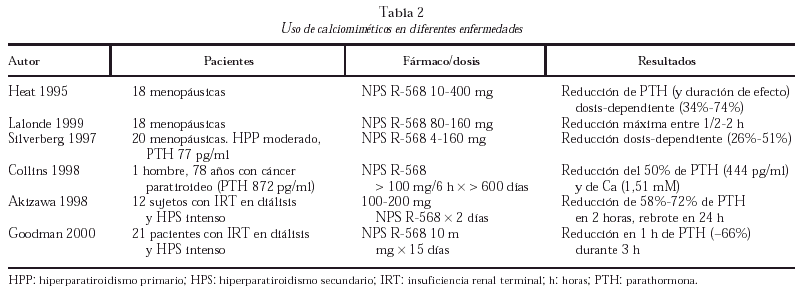
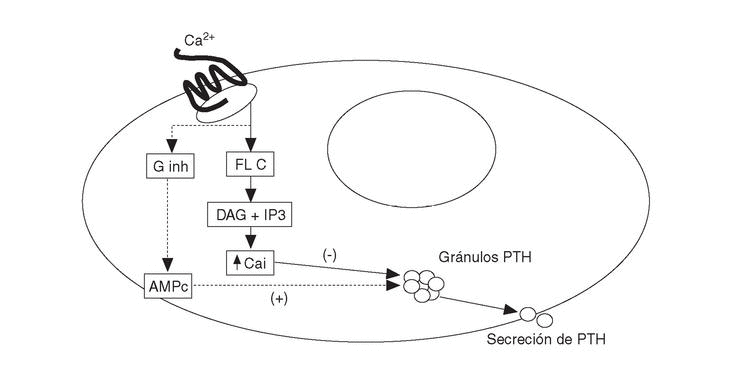
Los agonistas del RSCa activan las fosfolipasas A2, C y D en células paratiroideas bovinas. La activación de las fosfolipasas produce un aumento transitorio de Cai por el aumento concomitante de inositol trifosfato (IP3). Además inhibe una o más isoformas de la proteína G inhibitoria, determinando ambas acciones la reducción de la secreción de PTH en las células paratiroideas (fig. 1). El mecanismo íntimo por el que se produce la inhibición de la secreción de PTH (curiosamente el RSCa es activador en el resto de células) es desconocido. A pesar de que sus acciones en otras células son diferentes (tipo factor de crecimiento similar a la insulina-1 [IGF-I] o activando quinasas mitogénicas), actúa de forma similar en células exocrinas lo que sugiere un papel común en el control de las secreciones endocrinas y exocrinas.
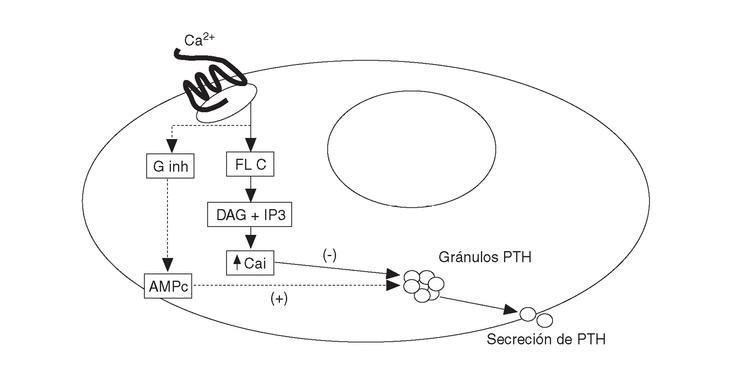
Fig. 1. Activación del receptor sensible a calcio (RSCa) en células paratiroides. Ginh: proteína G inhibitoria; FL C: fosfolipasa C; DAG: diacil-glicerol; IP3: inositol-trifosfato. PTH: parathormona; AMPc: monofosfato de adenosina cíclico; Ca: calcio iónico.
La expresión del RSCa se reduce en cultivo (> 80%), así como en los adenomas paratiroideos y en el fallo renal crónico, mientras que la (interleucina-1β) (IL-1β)y la vitamina D activa aumentan modestamente su expresión.
El 90% de los sujetos con hipercalcemia hipocalciúrica familiar (HHF) portan mutaciones heterocigotas inactivadoras del RSCa en células paratiroideas y renales (fig. 1). En esta enfermedad existe un set-point elevado para la inhibición de la secreción de PTH y una gran avidez por la reabsorción tubular de calcio por lo que, a diferencia del HPP, la calciuria es baja. La pérdida de función del RSCa en la HHF es variable alcanzando su máxima expresión en las mutaciones homocigotas (en un caso se ha descrito una mutación heterocigota compleja) que originan el hiperparatiroidismo neonatal intenso, fatal salvo paratiroidectomía en las primeras semanas de vida. En el extremo opuesto del espectro se encuentran la hipocalcemia autosómica dominante, caracterizada por una clínica similar al hipoparatiroidismo pero con mayor calciuria, en la que se han descrito mutaciones activadoras del RSCa6.
Parece razonable por lo expuesto hasta ahora, buscar alteraciones en la expresión del RSCa en el HPP. Ni los estudios indirectos ni la secuenciación directa han demostrado la existencia de mutaciones del RSCa en el HPP5, aunque sí se ha observado una reducción significativa de ARNm del RSCa (25%-60%) en las glándulas afectas. No obstante, la reducción de la expresión de RSCa no correlaciona con las características clínicas o anatomopatológicas7. Así, aunque el gen RSCa no está mutado ni ausente en el HPP, parece que la reducción de la concentración de RSCa en la superficie de las células de adenomas paratiroideos causada por factores no conocidos puede ser importante en la fisiopatología de la enfermedad. Recientemente se ha demostrado que el grado de reducción de expresión de RSCa sí correlaciona con la alteración del set-point de liberación de PTH y ésta, a su vez, se relaciona con algunas características clínicas del HPP como la calcemia o el volumen del adenoma8.
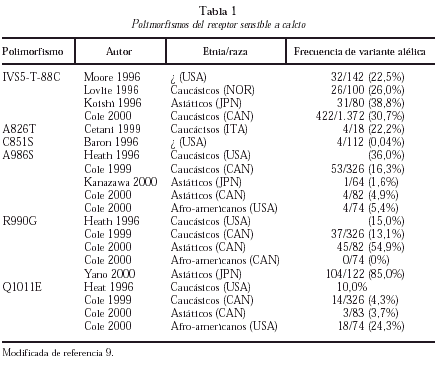
En los últimos años, se ha comunicado la existencia de diferentes polimorfismos en la región C-terminal del RSCa (tabla 1)9. El mejor estudiado es el polimorfismo A986S más frecuente en caucásicos y raro en asiáticos y negros. De diversos estudios de corte se deduce que este polimorfismo se asocia a hipercalcemia e hipocalciuria y a un mayor riesgo de HPP10. También han aparecido recientemente publicaciones que ligan los polimorfismos del RSCa a la calcemia en la población general o a la densidad mineral ósea en adolescentes, si bien se trata de estudios escasamente confirmados o con problemas metodológicos.
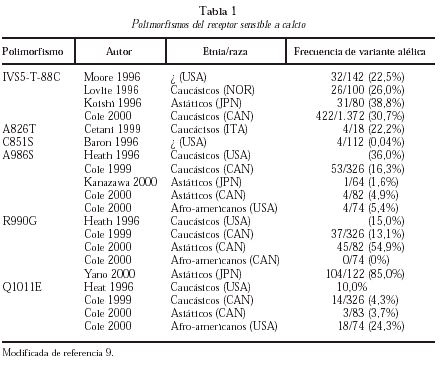
Así pues, a modo de resumen, puede decirse que en los pacientes con HPP no se han detectado mutaciones en el gen del RSCa, si bien en adenomas se ha descrito la reducción de la expresión de la banda del promotor de 5.4 kd del RSCa (exón 1A) de significación no totalmente esclarecida. En el HPP existe un aumento del 20%-30% en el set-point de liberación de PTH-Cai-dependiente, que se relaciona con la reducción variable de la densidad del mRNA y a RSCa en las glándulas paratiroideas patológicas, siendo más frecuente el desarrollo de HPP en los sujetos con alelo A986S.
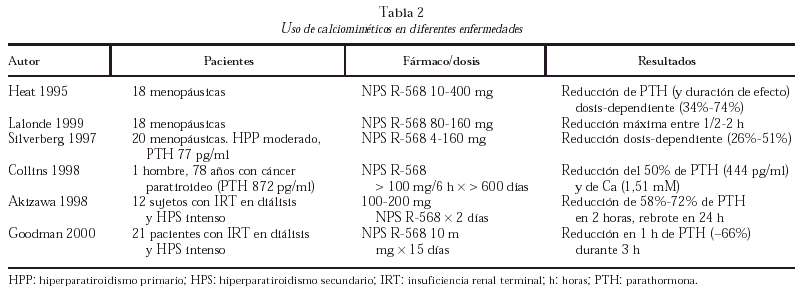
Las posibilidades terapéuticas de distintas moléculas capaces de ligar el RSCa son evidentes (tabla 2), especialmente en el tratamiento del HPP; se trata de fenil-alquil-aminas hidrofóbicas de bajo peso molecular capaces de inducir una modificación alostérica que activa el RSCa por unión a dominios transmembrana. Los mejor estudiados son NPS R-467 y NPS R-568; estas sustancias no son efectivas en ausencia de Cai o agonistas policatiónicos, son capaces de reducir la concentración de PTH en minutos en más del 50%, disminuyendo la calcemia en horas, y han demostrado inhibir la proliferación celular de las células paratiroideas. También se encuentran en desarrollo agonistas de corta acción del RSCa para el tratamiento de la osteoporosis por su capacidad de producir pulsos de PTH, así como antagonistas específicos renales que pueden ser de gran eficacia en las nefrolitiasis cálcicas recurrentes.