


Introducción
La capacidad de los fagocitos para generar radicales libres de oxígeno es parte esencial de los mecanismos de defensa contra infecciones en humanos. La producción de radicales libres en el estallido respiratorio es mediado por la enzima NADPH oxidasa.1 La importancia de la funcionalidad intacta del complejo de la NADPH oxidasa es demostrado clínicamente en los pacientes con Enfermedad Granulomatosa Crónica (EGC).2,3
La EGC es una inmunodeficiencia primaria se caracteriza por un defecto de fagocitosis, específicamente en el complejo enzimático de la NADPH oxidasa; tiene una incidencia estimada de uno en 250 000 recién nacidos vivos.4 Las infecciones y lesiones granulomatosas son las manifestaciones más frecuentes. El pulmón, piel, ganglios linfáticos e hígado son los sitios más frecuentes de localización de infecciones.5,6 La EGC confiere predisposición a infecciones por bacterias y hongos como Staphylococcus aureus y Aspergillus; además de otras causadas por micobacterias como el Bacilo Calmette-Guérin (Tabla 1).7
La NADPH oxidasa está constituida por seis subunidades: una cadena β glucosilada de 91 kDa (gp91phox [Phagocyte oxidase]), una cadena no glucosilada de 22 kDa (p22phox), y las proteínas p47phox, p67phox, p40phox y la enzima pequeña GTPasa p21rac (Rac1 en neutrófilos y Rac2 en macrófagos). Estas proteínas se encuentran, en sus formas inactivas, agrupadas en el complejo citocromo b558 (gp91phox + p22phox), asociadas con la membrana citoplásmica de los gránulos de los fagocitos y como proteínas citosólicas (p47phox, p67phox, p40phox y p21rac).8,9
Durante el proceso de la fagocitosis se inicia la señalización intracelular río abajo, los componentes citosólicos p47phox y p67phox se fosforilan y sufren cambios conformacionales que favorecen su unión a p40phox y a citocromo b558, activándose la NADPH oxidasa para así transferir electrones a moléculas de oxígeno y formar radicales libres de oxígeno (superóxido, peróxido de hidrógeno e hipoclorito), fenómeno conocido como "estallido respiratorio". Los radicales libres de oxígeno, son capaces de interactuar con el ion potasio y las enzimas lisosomales, que en conjunto destruyen proteínas, polisacáridos, lípidos, ADN y ARN de los microorganismos fagocitados.6,8,10
La EGC se origina por una alteración funcional en una de las subunidades de la NADPH oxidasa, debida a una mutación en cualquiera de los genes que las codifican. Hasta el momento se ha identificado en el patrón de herencia ligado al X, que hay una mutación en el gen CYBB este codifica para la proteína gp91phox con una frecuencia de 65% a 70% de los casos; y dentro de las autosómicas recesivas las mutaciones en los genes CYBA que codifica para la proteína p22phox (frecuencia de 5%); NCF1 que codifica para la proteína p47phox (frecuencia 25%); el NCF2 que codifica para la proteína p67phox (alrededor de 5%) y NCF4 que codifica para p40phox (menos de 1%).10-13
Es importante considerar que en 10% de los casos con EGC RLX existen mutaciones de novo, es decir que el paciente es el primer afectado en un pedigree, en estos casos el defecto genético no se trasmite de los progenitores, sino se genera en la célula germinal durante la embriogénesis.14
Patrones de transmisión hereditaria en EGC
La EGC es una enfermedad hereditaria con dos patrones de herencia. En el patrón autosómico recesivo ambos padres son portadores de la mutación en uno de sus dos alelos; la posibilidad teórica de que sus hijos sean portadores, como los padres es de 50%, mientras hay un riesgo de 25% para hijos afectados por la enfermedad, y otro 25% ni afectados ni portadores, totalmente sanos y libres.15
En el patrón de herencia recesiva ligada al cromosoma X los individuos afectados son del sexo masculino; esto se explica porque al tener la mujer dos cromosomas X, uno con un gen mutado y otro con un gen normal, el último va a librar de enfermedad; como los hombres sólo tienen un cromosoma X, este contendrá un gen mutado que causará la enfermedad. La probabilidad de descendencia afectada dependerá del sexo del progenitor que porta la mutación. Un hombre enfermo tendrá 100% de hijas portadoras y 100% de hijos sanos. Una mujer portadora tendrá 50% de sus hijas portadoras y 50% de hijos varones enfermos.15
Las portadoras en la EGC, importancia de su detección
La lionización es un fenómeno que tiene lugar durante la embriogénesis temprana y es irreversible, a través de éste, las células de un organismo con dos cromosomas X, (X1, X2), uno de ellos es inactivado y aparece en los llamados Corpúsculos de Barr. Forma parte de los mecanismos de compensación de dosis génica, evitándose que las mujeres expresen el doble de producto génico por disponer de dos cromosomas X (frente a un X y otro Y en los hombres). El cromosoma inactivado es diferente en cada célula, es decir que en unas células el que se inactiva es el X1 y en otras el X2, lo cual explica el mosaicismo en un individuo.16 El fenómeno de lionización en las mujeres portadoras de EGC RLX, explica que un porcentaje de los neutrófilos expresen el gen mutado (no produzcan radicales libres), y otro porcentaje de los neutrófilos expresen el gen normal (produzcan radiales libres). Como el fenómeno de lionización es aleatorio, el porcentaje de una población de neutrófilos con producción de radicales libres normal y de otra con producción de radicales libres anormal va variar de portadora a portadora de EGC.17 La mayoría de ellas tiene una población normal de neutrófilos de 10%, dicho porcentaje es suficiente para estar sin infecciones graves y recurrentes.18,19
Sin embargo, algunas de ellas presentan estomatitis aftosa recurrente y las lesiones cutáneas que clínica e histológicamente son parecidas a las del lupus discoide. Se han reportado casos raros de mujeres portadoras de EGC recesiva ligada al X que en edad adulta presentan infecciones bacterianas recurrentes o graves similares a la de los pacientes con EGC.20
Una vez que se confirma un nuevo caso de EGC se debe proceder a detectar que tipo de patrón de herencia se presenta en la familia, esto debido a que el asesoramiento genético que se ofrezca a la paciente y su familia será diferente para los casos con herencia AR que aquellos con herencia recesiva RLX. Como ya describimos previamente las portadoras de EGC RLX tienen un espectro clínico que va de ser asintomáticas, presentar algunas manifestaciones leves o presentar un fenotipo clínico similar a los pacientes con EGC.21
Métodos diagnósticos para pacientes y portadoras de EGC
Existen diversas técnicas de tamizaje para el diagnóstico de EGC, todas tienen en común la medición de los radicales libres producidos por los neutrófilos. Las técnicas más utilizadas en el laboratorio son la reducción de citocromo C, quimioluminiscencia, reducción de nitroazul de tetrazolio (NBT) y la oxidación de 1, 2, 3 dihidrorodamina (1, 2, 3 DHR).21,22 Cada una de estas técnicas apoya fuertemente el diagnóstico de EGC ante un cuadro clínico sugestivo. Algunas de ellas pueden ser útiles para la detección de portadoras de EGC RLX. Sin embargo, el estándar de oro para el diagnóstico de pacientes con EGC, tanto AR como RLX, lo mismo que para los portadores de EGC AR o portadoras de EGC RLX es la secuenciación directa de los genes candidatos.14,23
La reducción de NBT es la técnica que se utiliza con mayor frecuencia como tamizaje, es una técnica sencilla; consiste en colocar una gota de sangre del candidato en estudio en un portaobjetos, posteriormente se agrega PMA (forbol miristato acetato) con el fin de estimular a los neutrófilos; con esto hay una producción de radicales libres (superóxido), estos al reaccionar con el colorante NBT son reducidos a formazán; dicha reacción se hace evidente con un viraje de color amarillo pálido a azul pardusco en los neutrófilos sobre el porta objetos. Los neutrófilos de pacientes con EGC no producen radicales libres, por tanto muestran incapacidad para reducir NBT, observándose ausencia de cambio de color sobre el portaobjetos.24 Una de las desventajas de esta técnica es que a pesar de que es una prueba cuantitativa, el valor dado estará basado en el análisis subjetivo del observador. Es por esto que puede haber falsos negativos, principalmente en aquellos casos con afección en moléculas de la NADPH oxidasa como p22phox, p47phox, y p67phox, en los que existe una producción parcial de radicales libres. Además la detección de portadoras por está técnica tiene un margen amplio de error.25
Otros métodos útiles en la medición cuantitativa del estallido respiratorio son a través de la quimioluminiscencia y la reducción de citocromo c, debido a que requieren la separación de los neutrófilos de sangre periférica, los lleva a ser complicados y poco prácticos como métodos de tamizaje diagnósticos. La reducción de citocromo c consiste en incubar los neutrófilos con PMA, posteriormente medir la producción de superóxido a través de un cambio colorimétrico, el cual no se observa en ausencia del sistema de NADPH oxidasa funcional; este método no es útil para la detección de portadoras.26
Otras técnicas son aquellas basadas en métodos en la fluorescencia, en éstas se utilizan compuestos químicos que se hacen fluorescentes al tener contacto con los radicales libres producidos por los neutrófilos. La fluorescencia emitida es cuantificada por a través del citómetro de flujo. Los dos fluorocromos más utilizados son la diclorofluoresceina (DCF) y la 1, 2, 3 DHR.27,28
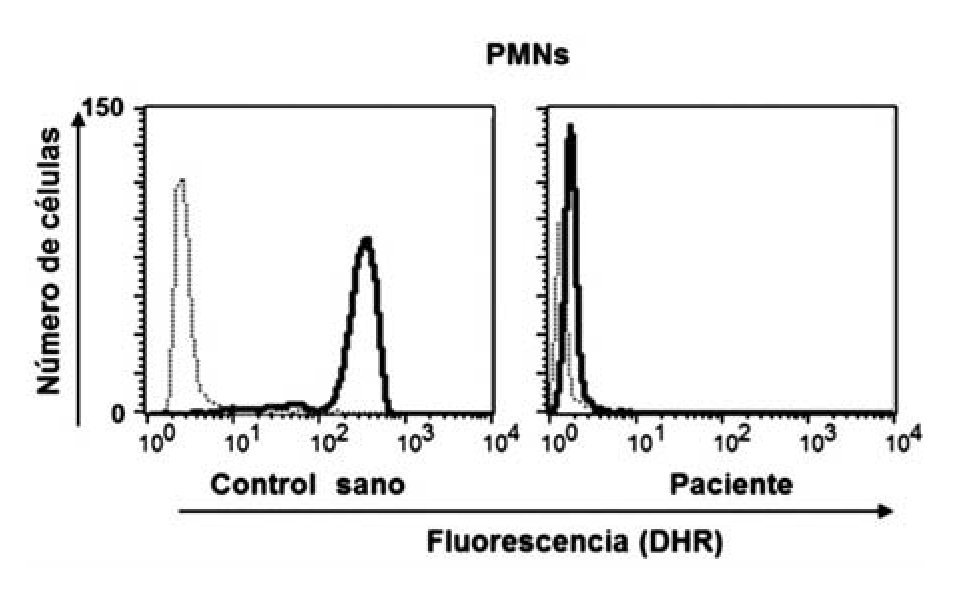
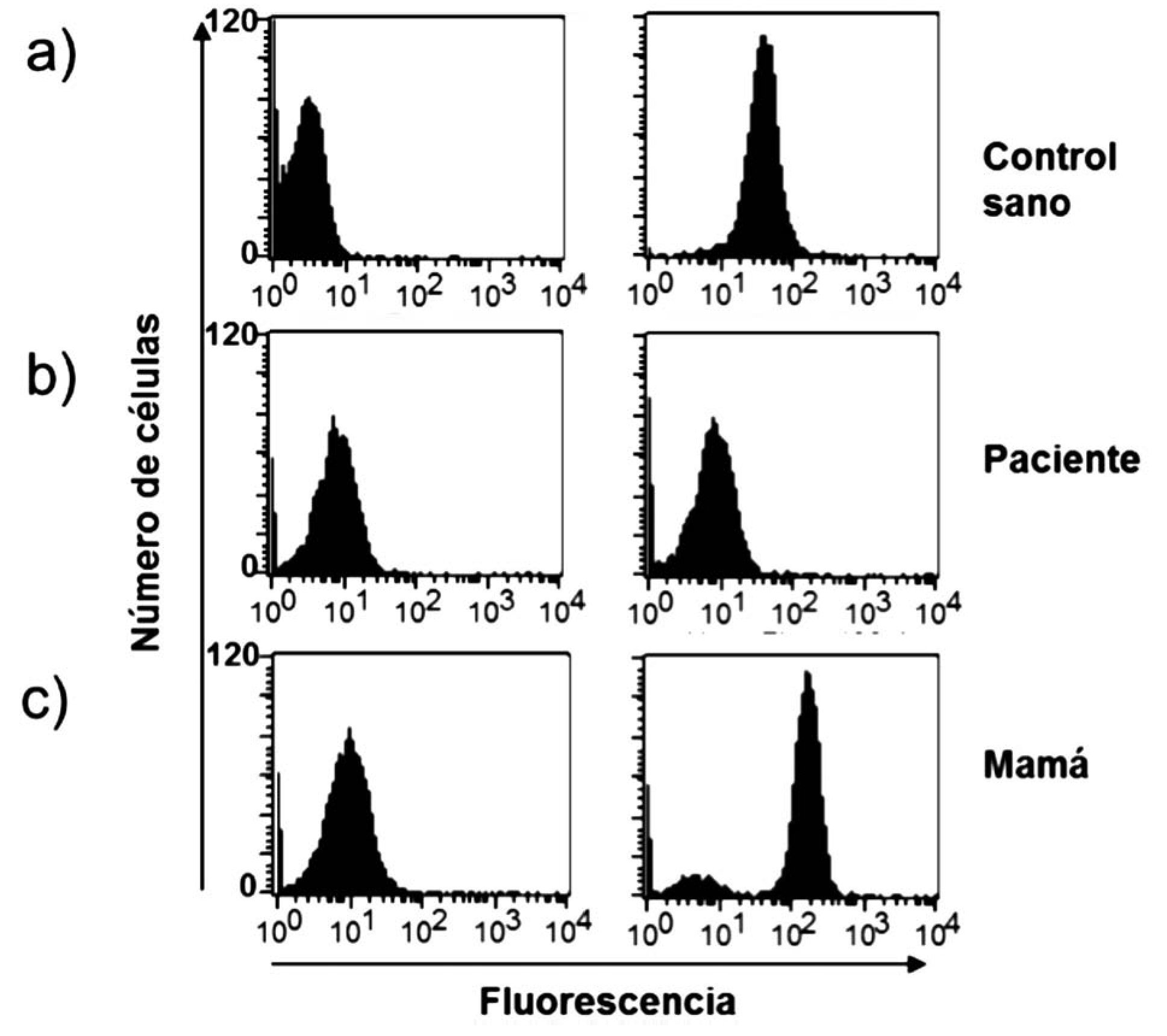
Dentro de las ventajas de la técnica de la reducción de 1, 2, 3, DHR están que se realiza en sangre total, sólo se requieren 0.1 mL (ideal para neonatos y lactantes) y es rápida; además de que es posible detectar la presencia de portadoras de EGC RLX a través del patrón de histograma y por tanto el patrón de transmisión hereditario. El fundamento teórico de a técnica de reducción de 1, 2, 3, DHR se basa en que la dihidrorodamina (no fluorescente) atraviesa la membrana plasmática de los neutrófilos, una vez dentro reacciona con el peroxido de hidrógeno para formar 1, 2, 3 rodamina (fluorescente). La conversión de la molécula no fluorescente a fluorescente depende completamente de la producción de los radicales libres (Figura 1). Dentro de los resultados obtenidos por la reducción de 1, 2, 3, DHR por citometría de flujo podemos tener tres posibilidades, sujetos sanos (en los que la población total de neutrófilos emite fluorescencia) (Figura 2a), pacientes con EGC (en los que la población total de neutrófilos no emite fluorescencia) (Figura 2b), portadoras de EGC RLX (en las que existen dos poblaciones de neutrófilos, una que emite fluorescencia y otra que no emite fluorescencia) (Figura 2c).29,30-33
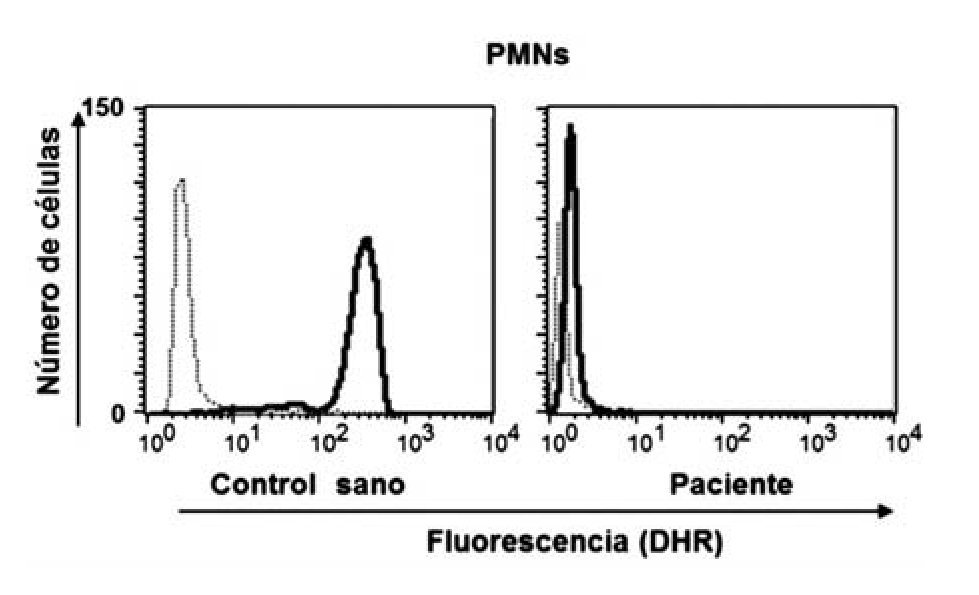
◊ Figura 1. Producción de radicales libres en polimorfonucleares (PMNs), con y sin estímulo, en un control sano y en un paciente con EGC, medición por oxidación de DHR.
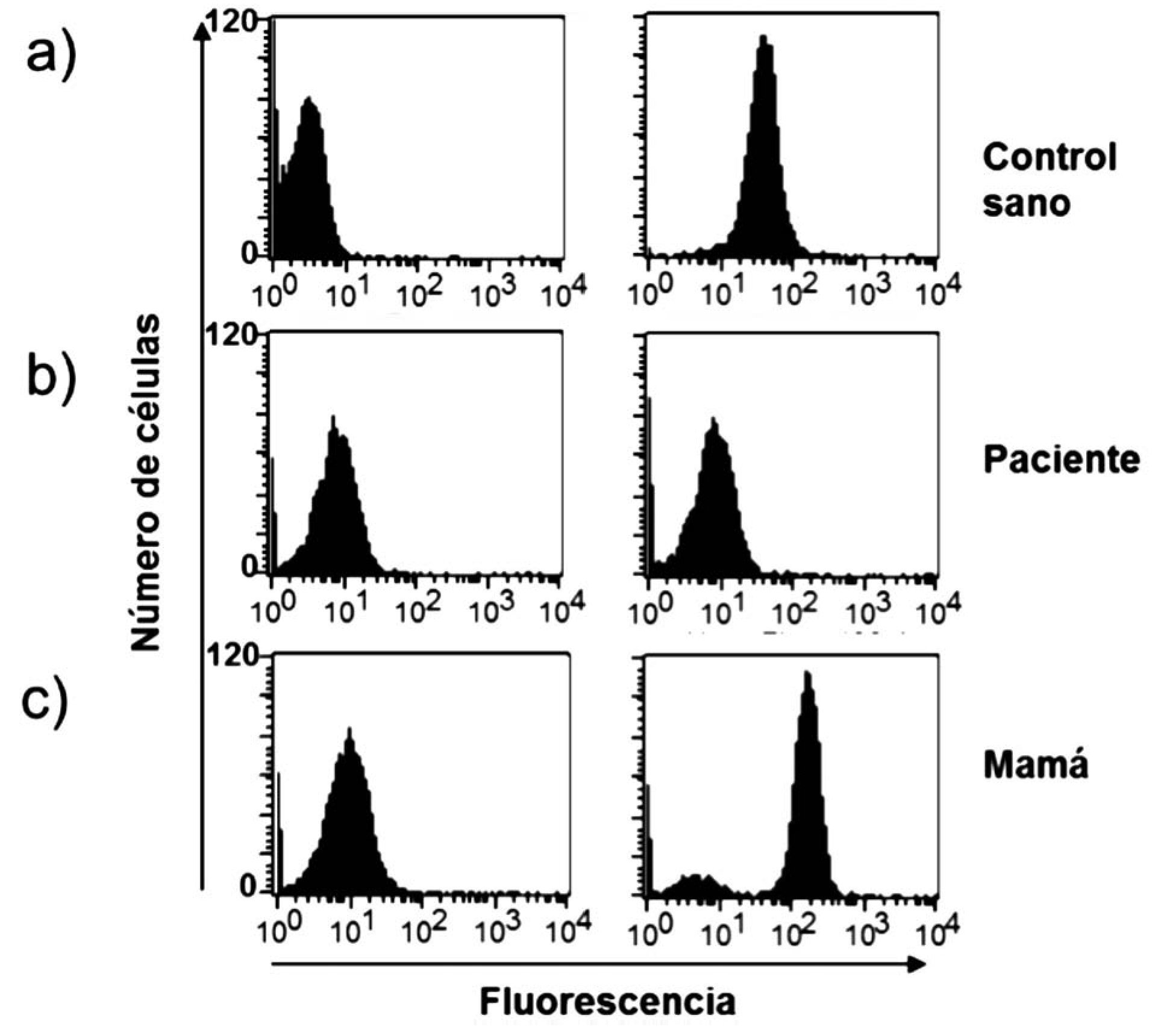
◊ Figura 2. Oxidación de DHR en polimorfonucleares (PMN), con y sin estimulo, en un control sano, en un paciente con EGC y en la mamá del paciente. Observe como el paciente muestra nula fluorescencia (b) y la madre presenta dos curvas debido a sus dos poblaciones de neutrófilos, una sana y otra con nula fluorescencia (c).
A pesar de que la mayoría de las diferentes técnicas de tamizaje para la EGC se realizan desde los años noventa, no existen estudios sistemáticos que describan la sensibilidad y especificidad de cada una de ellas. Richardson y colaboradores compararon a la reducción de DHR contra la prueba de reducción de NBT cuantitativa; describieron que ambas técnicas son equiparables en cuanto sensibilidad en la medición de radicales libres. La ventaja de la primera es que es más rápida y requiere menos cantidad de sangre.26
Conclusiones
La EGC es una inmunodeficiencia primaria que puede heredarse de dos formas autosómica recesiva y recesiva ligada al X, hasta el momento existen varios métodos diagnósticos para identificar la EGC. Sin embargo, es de vital importancia no sólo diagnosticar a los pacientes con EGC, sino también poder identificar su patrón de herencia, para brindar asesoramiento genético y además para orientar a las portadoras con respecto a las posible manifestaciones clínicas que pueden presentar a lo largo de su vida, como es el desarrollo de enfermedades autoinmunes. La conversión de 1, 2, 3 DHR a 1, 2, 3 rodamina mediante citometría de flujo es un ensayo rápido y sensible para el diagnóstico de EGC y puede ser una herramienta útil para identificar los patrones de herencia. Actualmente en la Unidad de Investigación en Inmunodeficiencias del Instituto Nacional de Pediatría, se realiza la determinación de 1, 2, 3 DHR para diagnóstico de pacientes con EGC y detección de portadoras.
Correspondencia: Dra. Lizbeth Blancas Galicia.
Insurgentes Sur 3700-C col. Insurgentes-Cuicuilco CP. 04530 Coyoacán, México Distrito, Federal.
Teléfono: 1084 0900, ext. 1866
Correo electrónico:lbg73_2000@yahoo.com.