



Introducción
El angioedema hereditario (AH) es una enfermedad poco frecuente, pero de gran impacto sobre la calidad de vida del paciente, tanto por la recurrencia de los síntomas, como por el riesgo para la vida en ciertas circunstancias.
Es una enfermedad de origen genético, transmitida con un patrón autosómico dominante. Se caracteriza por la presencia de episodios recurrentes de angioedema no pruriginoso que puede afectar cualquier parte del cuerpo; típicamente involucra las extremidades inferiores y superiores, el aparato digestivo, la cara y la vía respiratoria.1,2
Antecedentes históricos
La primera descripción de la enfermedad se ubica en 1586 cuando Donati describió pacientes con angioedema, sin asociación con urticaria. En 1883 Quincke informó casos de la enfermedad, llamándola edema angioneurótico.1-3 Correspondió a Sir William Osler, en 1888, el comentar el carácter hereditario de la enfermedad y su patrón autosómico dominante.1 La mejoría en las técnicas de laboratorio permitió a Virginia Donaldson identificar el defecto molecular del Inhibidor de C1 en 1963.3
Epidemiología
La prevalencia de esta enfermedad, con base en la bibliografía, se ha determinado en 1:10 000 y hasta 1:50 000, sin encontrar diferencias de género ni predominio de raza.1 No existen datos con respecto a la población mexicana. En el servicio de Alergia e Inmunología Clínica del Hospital General de México, en los últimos cinco años se ha manejado un paciente. En la bibliografía nacional se describen siete casos, publicados en la Revista de Alergia.4,5 La enfermedad inicia en la infancia, alrededor de los dos a tres años de edad.1 Más de la mitad de los casos presenta síntomas a los 10 años. Se ha observado que la enfermedad empeora durante la adolescencia y persiste a lo largo de la vida, con un curso impredecible.
Genética
El AH es resultado de mutaciones en el gen del Inhibidor de C1 localizado en el cromosoma 11 (p11.2-q13).6 Los estudios de genética han identificado más de 150 diferentes mutaciones sin encontrar hasta el momento una correlación clara entre el defecto genético y la severidad de la enfermedad.1 Si bien se han relacionado mutaciones que producen proteínas truncadas o alteración en el procesamiento de la proteína y que se relacionan con el tipo I. Las mutaciones en el sitio activo, generalmente en el exón 8, se relacionan con una proteína defectuosa que se produce en cantidad normal o aumentada, lo cual se encuentra en el tipo II.6
Debe considerarse que a pesar de identificarse un patrón de herencia autosómico dominante, en una cuarta parte de los pacientes no se tendrá el antecedente familiar de la enfermedad por tratarse de mutaciones espontáneas.1
Clasificación
El AH se ha dividido en tres tipos, dos de los cuales son atribuibles a mutaciones en el inhibidor de C1 (IC1) y son indistinguibles clínicamente.2
El Tipo I se presenta en 85% de los casos. Se caracteriza por niveles disminuidos de IC1 con alteración de su función.6
El Tipo II representa 25% de los casos. Los niveles del IC1 pueden ser normales o incluso encontrarse elevado, pero su función es anormal.5
El tipo III representa 1% de los casos. Los pacientes tienen niveles normales del IC1, la mayoría son del género femenino, con pocas excepciones. La administración de estrógenos se relaciona con el inicio o exacerbación de los síntomas y pueden relacionarse con el ciclo menstrual. El defecto genético se ha identificado en el gen del Factor XII de la coagulación.6
Fisiopatología
Inhibidor de C1: El inhibidor de C1 es un miembro de la familia de serpinas o inhibidores de serin proteasa, también conocido como SERPING1.7 Es una proteína sintetizada en el hígado y representa el principal inhibidor de las proteasas del complemento (C1r, C1s, MASP-1 y MASP-2) y de las proteasas del sistema de contacto. También es un inhibidor menor de las proteasas del sistema fibrinolítico, y de la coagulación.6-8
Los pacientes con angioedema hereditario son heterocigotos para el gen del inhibidor, es decir, sólo esta afectado uno de los alelos. Por tanto, se esperarían niveles del inhibidor de por lo menos 50% de su valor normal. Los niveles encontrados en estos pacientes son menores, entre 30% y 50%.
Esto se atribuye al mecanismo de acción de las serpinas, las cuales forman un complejo serpinaproteasa en la que ambas se inactivan y son degradadas. Al consumirse el IC1 en las distintas vías en las que participa, se consume, presentándose niveles inferiores a lo esperado.7
En pacientes con el tipo II de la enfermedad, los niveles son normales o elevados ya que por el defecto en la función de la proteína, se prolonga su vida media al no poder formar complejos enzimáticos.7
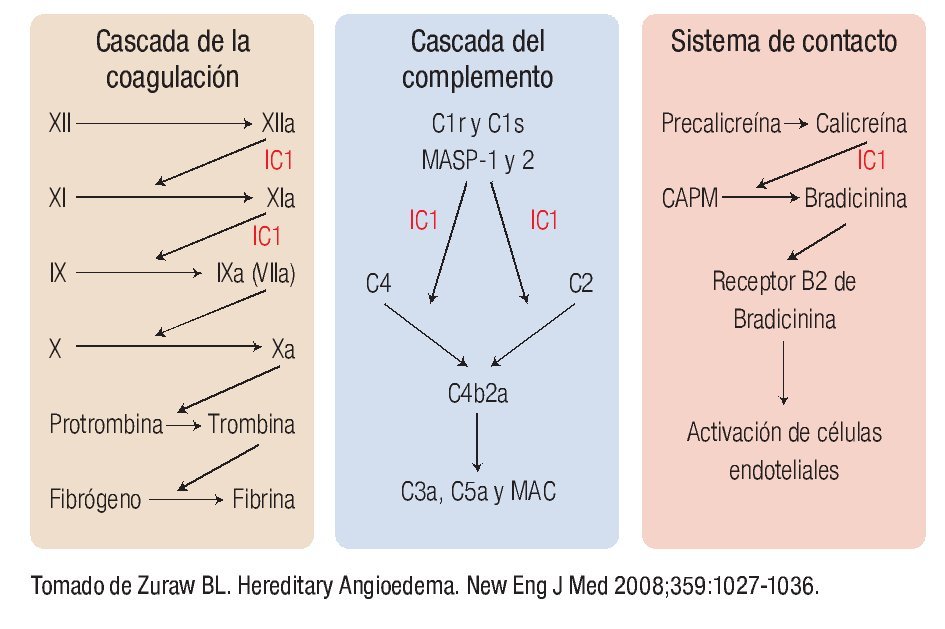
En la Figura 1 se muestra la cascada de la coagulación, la del complemento y el sistema de contacto, así como los sitios en los que interviene el IC1. Sus principales objetivos son el factor XIIa y XIa de la coagulación, C1r, C1s, MASP-1 y 2 de la vía clásica y de las lectinas respectivamente y la calicreína, importante mediador responsable de la generación de bradicinina a partir del cininógeno de alto peso molecular.1,8 Como veremos más adelante, el principal mediador responsable de la generación del angioedema es la bradicinina. Es importante recordar que debido a la ausencia del inhibidor, se activa la cascada del complemento, consumiéndose C2 y C4. En la vía fibrinolítica también interviene al impedir la conversión de plasminógeno en plasmina. Sin embargo, su papel en esta vía es de menor relevancia al intervenir otros mediadores en su regulación.1,7,8
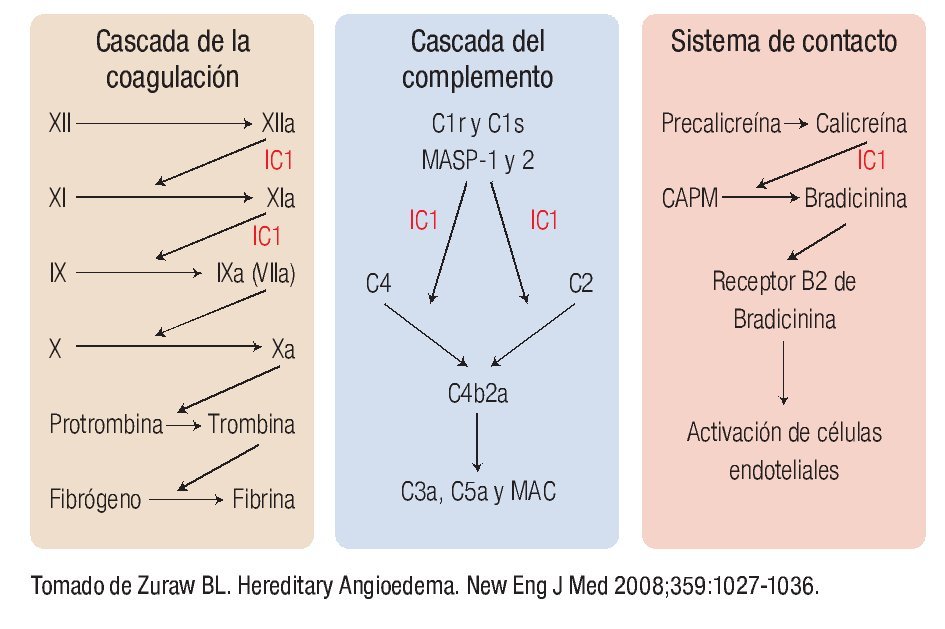
◊ Figura 1. Cascada de la coagulación, del complemento y sistema de contacto y los sitios de acción del inhibidor de C1.
Bradicinina: Las cininas son péptidos que estimulan la relajación del músculo liso vascular y aumenta la permeabilidad vascular. De interés en esta patología es la bradicinina, producto de la activación del sistema de contacto.7 Se ha identificado como el principal mediador responsable del angioedema hereditario. Esta cinina ejerce su acción biológica principalmente a través de su receptor B2. Éste se expresa de manera constitutiva en las células del endotelio vascular y en las células del músculo liso.6-8,10
En los humanos existe otro receptor, el B1, expresado en los tejidos posteriores a la lesión o al contacto con endotoxinas bacterianas. Este receptor podría participar en la generación de angioedema y explicar la relación del cuadro clínico con factores como el trauma físico o las infecciones. Esto todavía se encuentra en estudio.7
La degradación de la bradicinina se lleva a cabo por medio se cininasa I y II, conocidas como carboxipeptidasa N y ECA. Esto explica por el que el uso de antihipertensivos de tipo inhibidores de la enzima convertidora de angiotensina (IECA) exacerban los síntomas en estos pacientes.6-8
Cuadro clínico
Se caracteriza por síntomas prodrómicos hasta en un tercio de los casos e incluyen parestesias y eritema marginado, un rash serpinginoso no pruriginoso.2
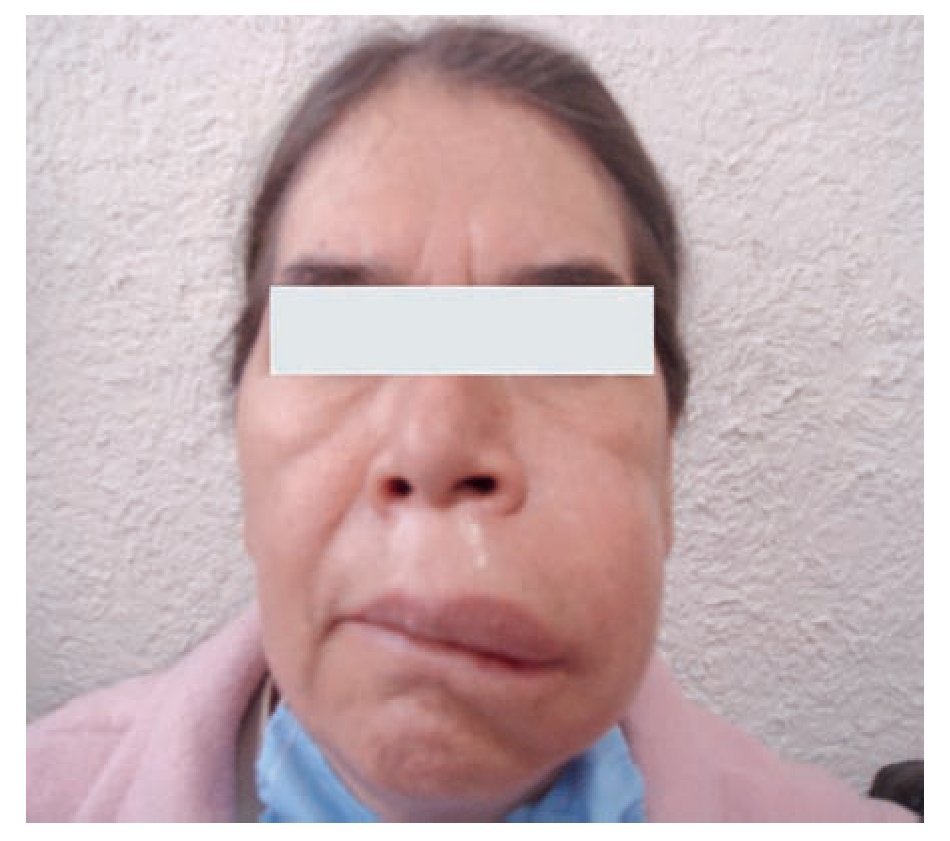
Típicamente, el edema se desarrolla durante 24 horas y cede durante las 48 a 72 horas siguientes. Las zonas más afectadas son cara, brazos, piernas, manos, pies y abdomen,1 como se muestra en la Figura 2. Puede presentarse en una región corporal, para luego migrar a otra antes de resolverse o incluso, afectar múltiples sitios al mismo tiempo. Es importante mencionar que la ausencia de urticaria es una característica de esta enfermedad.
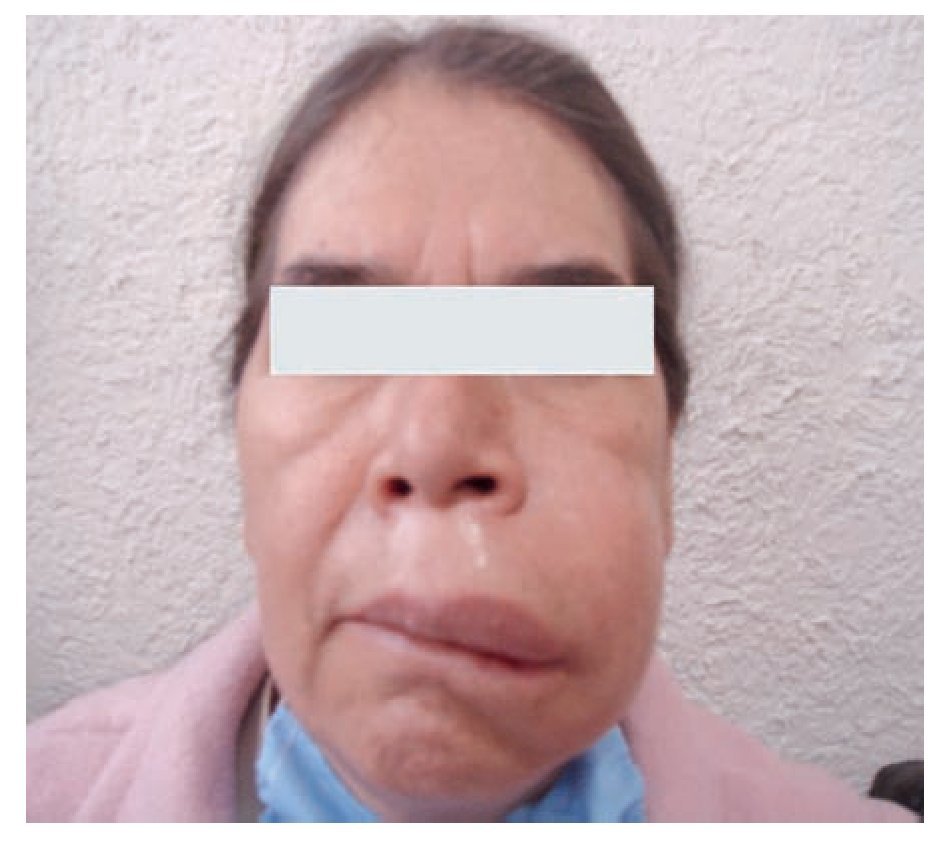
◊ Figura 2.Paciente de la consulta externa con Angioedema Hereditario del Servicio de Alergia e Inmunología Clínica. Hospital General de México.
Más de 90% de los pacientes presenta un episodio abdominal en alguna ocasión.10 El dolor abdominal es intenso, acompañado de diarrea (41%), náusea y vómito (71%). Los ruidos peristálticos pueden estar disminuidos o ausentes y tener datos de irritación peritoneal, es decir, simular un abdomen agudo. Durante los episodios abdominales existe un paso de líquido intersticial hacia la cavidad peritoneal, lo que puede ocasionar hipotensión. Si afecta el aparato digestivo representa un reto diagnóstico ya que un tercio de los pacientes pueden ser sometidos a procedimientos quirúrgicos innecesarios, sobre todo en quienes no se ha realizado el diagnóstico de la enfermedad.
El edema laríngeo representa un riesgo para la vida del paciente. Es menos común; sin embargo, 50% de los pacientes presentan un episodio de este tipo alguna vez en su vida. Se reporta que 30% de las muertes en pacientes con AH se deben a edema laríngeo y asfixia.1,9
Factores desencadenantes
Entre los factores que se han identificados como desencadenantes de los síntomas se incluyen: el trauma físico, procedimientos quirúrgicos, médicos y dentales, bipedestación prolongada, infecciones, estrés emocional y algunos fármacos.1,2,8 Estas situaciones deben evitarse o hacer ajustes en el tratamiento como se describirá más adelante.
Diagnóstico
El retraso en el diagnóstico de la enfermedad es común debido a la falta de sospecha del padecimiento. Hace 33 años, el tiempo promedio entre el inicio del cuadro clínico y el diagnóstico era de 22 años, mientras que hace cinco años tardaba 10 años.1 Hoy la medicina ayuda al diagnóstico temprano y oportuno. Se cuenta con criterios clínicos y de laboratorio para hacer el diagnóstico de AH, los cuales se presentan en la Tabla 1. Es necesario que se cuente con un criterio clínico y uno de laboratorio.

Además de los criterios mencionados previamente, se deben determinar los niveles plasmáticos de C4, ya que por la deficiencia del IC1, se consumen otros factores del complemento. Concentraciones menores a 30% aparecen en 90% de los pacientes, causando duda en cuanto al diagnóstico si los niveles son normales durante un episodio agudo.19,11
Diagnóstico diferencial
Deben considerase otras causas de angioedema como las de etiología alérgica; enfermedades autoinmunes con presencia de edema facial como lupus eritematoso sistémico, polimiositis, dermatomiositis; enfermedades tiroideas; síndrome de vena cava; queilitis granulomatosa y enfermedad de Melkersson-Rosenthal; y triquinosis.12
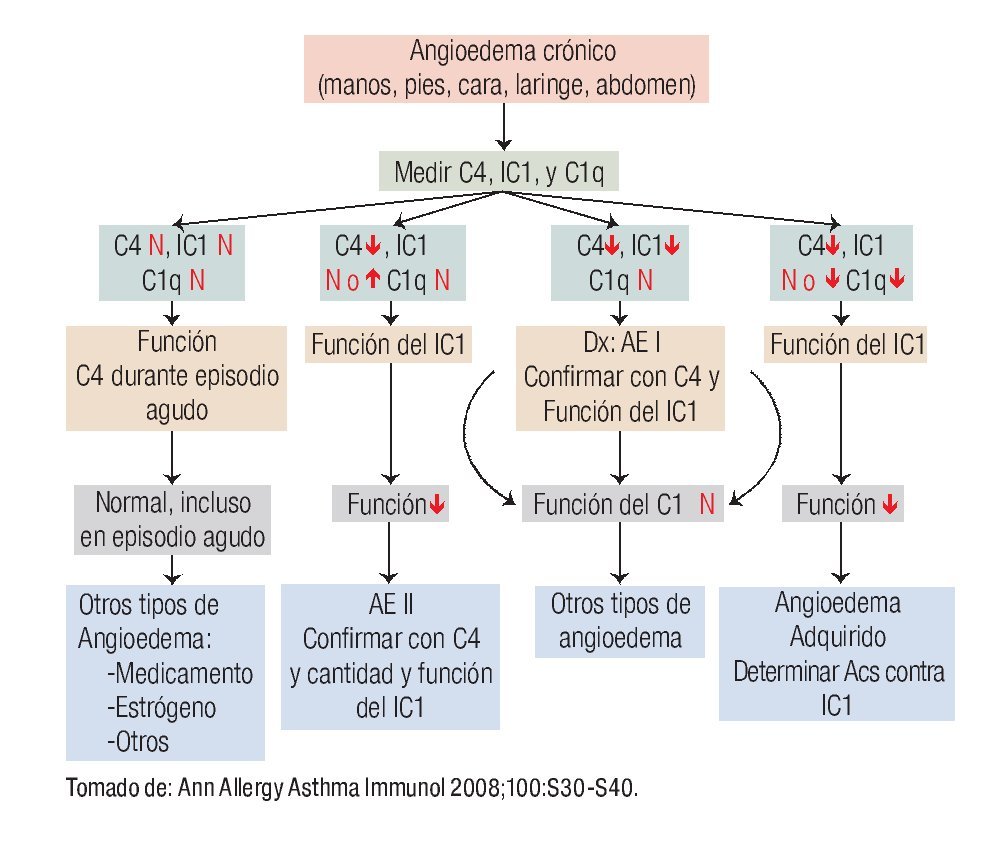
En la Figura 3 se muestra el algoritmo diagnóstico para confirmar diagnóstico de los diversos tipos AE, los estudios de laboratorio que se sugieren y el diagnóstico diferencial con otros tipos de angioedema.
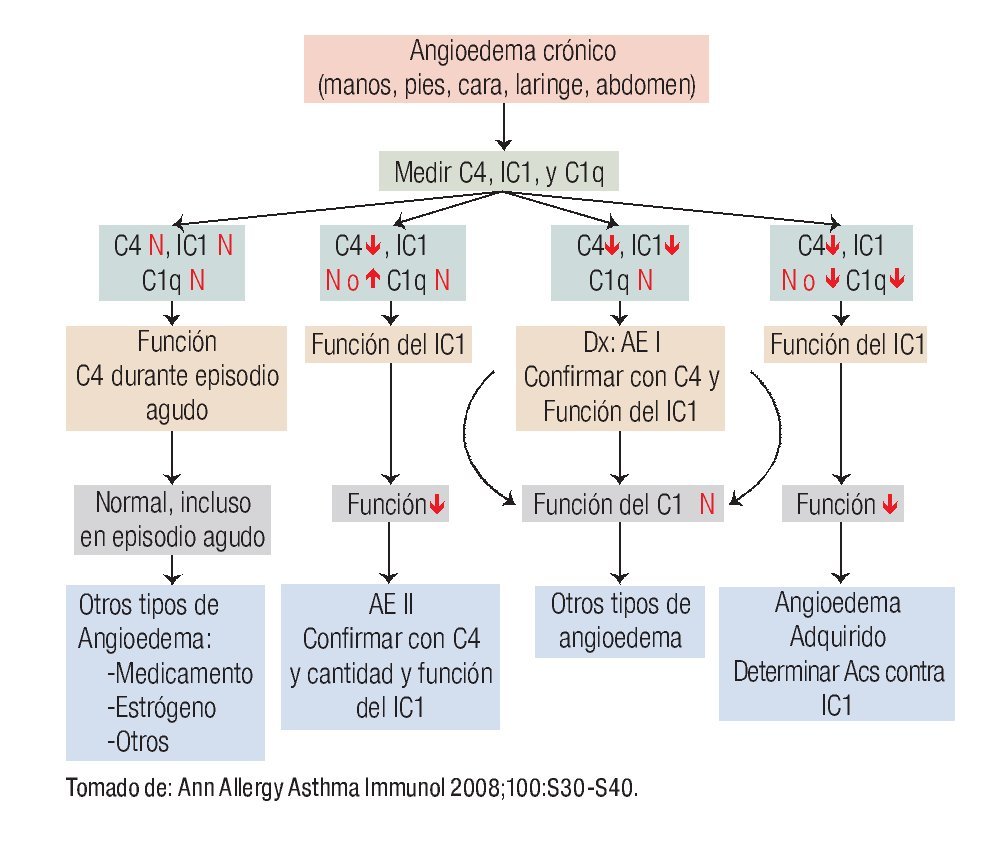
◊ Figura 3. Algoritmo diagnóstico del angioedema hereditario. ↑: aumento; ↓: disminución, N: normal, IC1: inhibidor de C1, Acs: anticuerpos.
Tratamiento
En los pacientes con angioedema hereditario consiste en:
1. Medidas generales: Dentro de las medidas generales es esencial determinar el factor desencadenante y evitarlo. En pacientes con riesgo de edema laríngeo es importante vigilar la permeabilidad de la vía aérea. No se recomienda la laringoscopia con el propósito de determinar la presencia de edema laríngeo ya que este procedimiento puede iniciarlo. El uso de analgésicos de tipo opioide puede ser necesario dada la intensidad del dolor abdominal, se indican también antieméticos. La reposición de volumen será necesaria en el caso de hipotensión.13
2. Profilaxis a corto plazo (PCP): Se considera este tipo de profilaxis ante circunstancias en las que existe riesgo de desarrollar el cuadro clínico e incluyen procedimientos dentales, estudios invasivos, cirugías, sobre todo aquellas en las que se tenga previsto realizar una intubación.1,14,15
3. Profilaxis a largo plazo (PLP): Se deben considerar aquellos pacientes en quienes la frecuencia o intensidad de los síntomas, afectan su calidad de vida. También los casos con más de un episodio agudo por mes. El edema laríngeo también es una indicación, así como la dependencia de analgésicos opioides debido a la intensidad y frecuencia del dolor abdominal. Pacientes con acceso limitado a los servicios de salud, pueden beneficiarse de la profilaxis a largo plazo sobre todo si presentan edema laríngeo.1
Tratamiento farmacológico
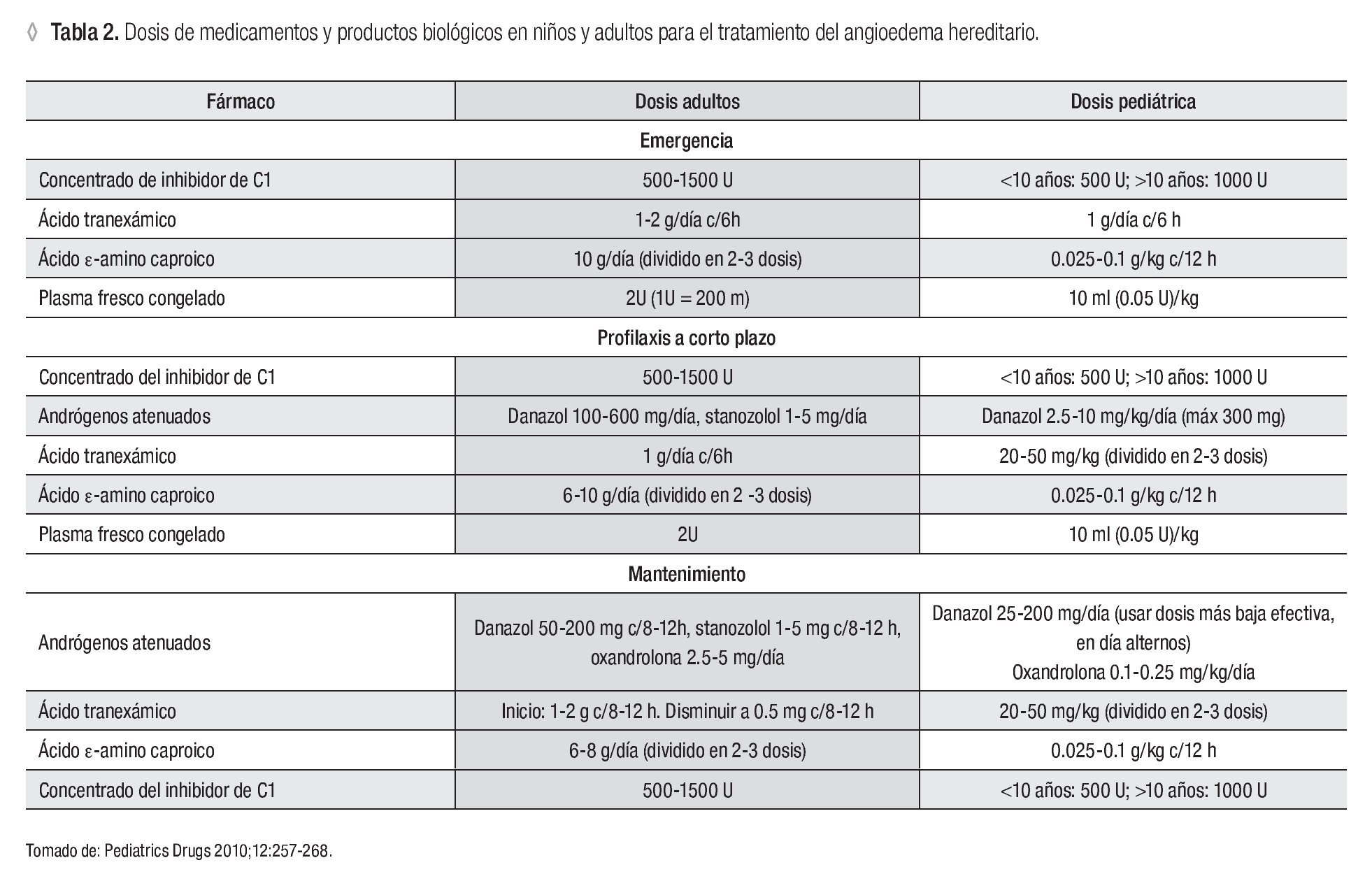
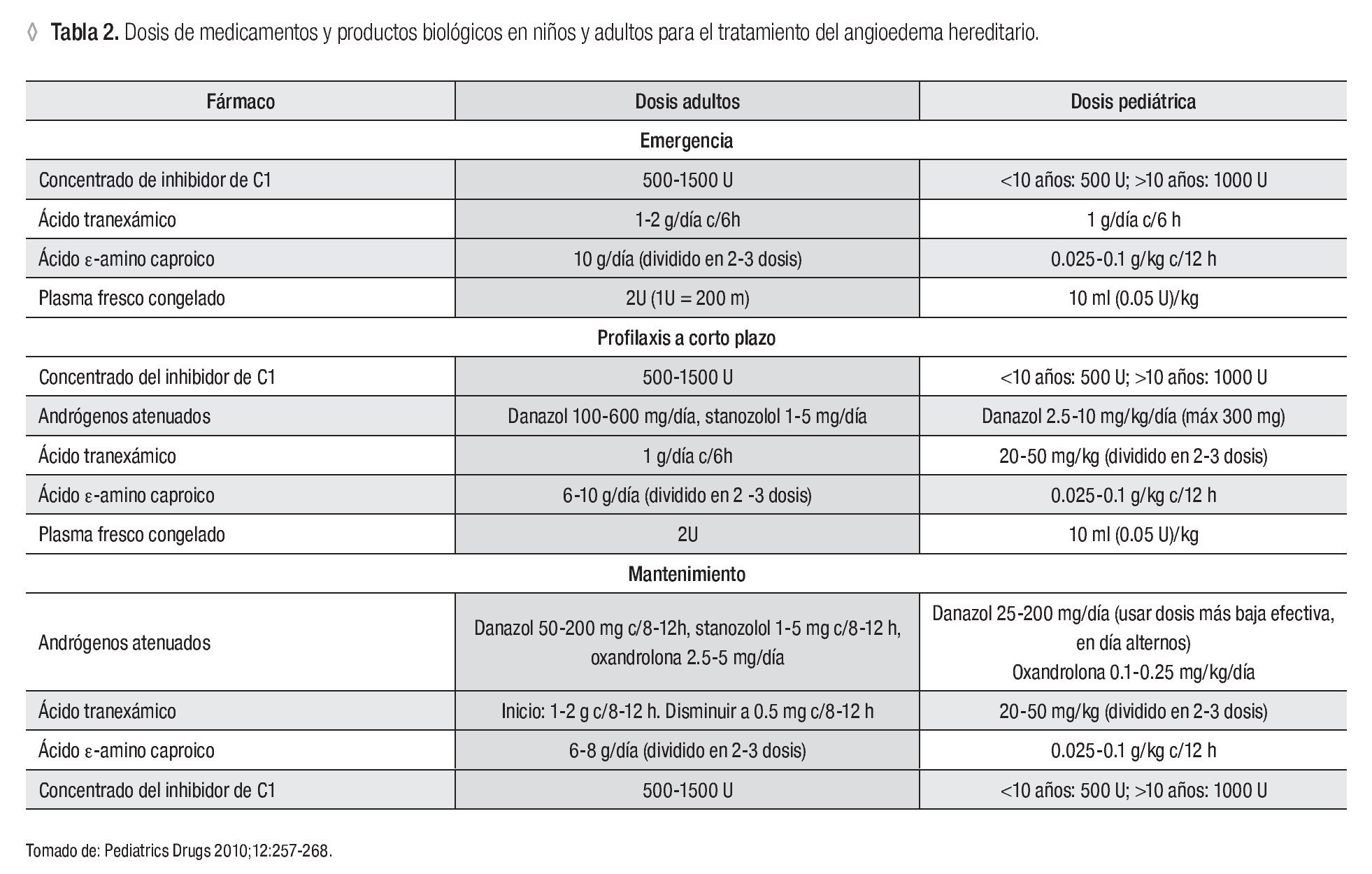
Existen diversas opciones terapéuticas para el tratamiento del AH. A continuación se comentará la evidencia con la que se cuenta para el uso de cada una de ellas. En la Tabla 2 se muestran las dosis pediátricas y del adulto para cada uno de los fármacos o biológicos recomendados.
Plasma fresco congelado (PFC): Con experiencia anecdótica se ha utilizado en los episodios agudos al reemplazar al C1-INH. La evidencia consiste únicamente en reportes de casos por lo que su uso es controversial. Existe el riesgo de que el PFC pueda empeorar el angioedema al reponer proteasas plasmáticas y los sustratos involucrados en la generación de péptidos relacionados con el angioedema. En los informes de casos se ha encontrado mejoría en 96% de los pacientes, utilizándose tanto en el episodio agudo como en la profilaxis a corto plazo.16
Andrógenos atenuados: Los andrógenos 17-α-alquilados, como el danazol, stanozolol y oxandrolona, se utilizan ampliamente en el tratamiento de esta enfermedad. Su utilidad en el tratamiento del AH se debe a que aumenta la producción del C1-INH a nivel hepático y de enzimas implicadas en la degradación de la bradicinina.16
Se indican en la profilaxis a corto y largo plazo y no en el episodio agudo por requerir varios días para ejercer su efecto.1,17
Los efectos adversos consisten en virilización y hepatotoxicidad. Se recomienda la monitorización de enzimas hepáticas cada seis meses y un ultrasonido hepático una vez al año mientras se estén utilizando.16,17
La valoración de la eficacia del tratamiento debe valorarse con la mejoría clínica, no con el aumento de los niveles del inhibidor o del complemento.
Debe utilizarse la dosis más baja que proporcione beneficios clínicos con el propósito de disminuir los efectos adversos.17 Se ha demostrado que los andrógenos atenuados proporcionan un beneficio al evitar los episodios agudos en 80% de los casos, independientemente de su localización, ya sea periférica o incluso edema laríngeo, disminuyendo éste último hasta en 95%.17
Antifibrinolíticos: Inhiben la activación del plasminógeno, evitando el consumo de C1-INH. Disminuyen el número y la severidad de los ataques, con menor efectividad que los andrógenos atenuados. Por tanto, se reservan para casos en los que no está indicado el uso de andrógenos atenuados. Su indicación es en la PCP y PLP.19 Sus efectos secundarios son dependientes de la dosis, entre los que se incluye un aumento en el riesgo de neoplasias en retina con el uso a largo plazo, indicándose una valoración oftalmológica.19,20
Concentrado del inhibidor de C1: La eficacia del uso de concentrado de inhibidor derivado del plasma se ha confirmado desde hace más de 25 años. Múltiples estudios han convertido al concentrado del inhibidor en el tratamiento de elección para los episodios agudos en los países en los que se encuentra disponible.
La infusión del concentrado del inhibidor lleva a un aumento en los niveles plasmáticos del mismo en 30 a 60 minutos y un aumento tardío en los niveles de C4. La efectividad del concentrado se ha demostrado para cualquier localización del angioedema incluyendo el edema laríngeo en cuyo caso puede salvar la vida del paciente. 21-23-25
Además de su utilidad en los episodios agudos, el concentrado del inhibidor se ha utilizado en la profilaxis a corto y largo plazo en aquellos pacientes con falla en la respuesta a los andrógenos atenuados o antifibrinolíticos.21
Existen tres tipos de concentrado del inhibidor, mencionados a continuación.
1. Purificado del plasma: Berinet es un liofilizado pasteurizado del inhibidor plasmático para su uso IV el cual ha demostrado su utilidad y a pesar de utilizarse durante más de 20 años en Europa, se aprobó por la FDA en el 2009. Su vida media es de 32 a 46 horas, no se informan recurrencias del angioedema debido a su vida media prolongada.21 Entre los efectos adversos que se reportan con su uso se encuentran: cefalea, fiebre, dolor abdominal, diarrea y vómito. En los estudios realizados se ha concluido que es efectivo a una dosis de 20 U/kg para el tratamiento de angioedema facial y abdominal comparado con placebo. A pesar de que no hay estudios doble ciego en los que se evalúe su efectividad en el tratamiento del edema laríngeo, en IMPACT-2 (estudio abierto) sugiere que es efectivo para el control del edema en esta localización.24
2. Nanofiltrado: Cinryze es un concentrado nanofiltrado pasteurizado para uso IV. Su producción es similar al concentrado derivado del plasma, pero se somete a un proceso de nanofiltración con el propósito de eliminar las partículas virales con o sin envoltura y posiblemente, los priones. Se aprobó en el 2008 por la FDA para su uso en adolescentes y adultos en la profilaxis de esta enfermedad. No se ha aprobado en episodios agudos.21 Se han realizado estudios en los cuales se demuestra su seguridad y eficacia en el tratamiento de ataques moderados a severos en los que involucra la cara, el abdomen y el aparato genitourinario. Su eficacia es independiente de la localización del angioedema.24
3. Recombinante: Rhucin es un C1-INH recombinante para su aplicación IV en episodios agudos producido en conejos transgénicos. Es idéntica a la proteína derivada del plasma y tiene la misma capacidad inhibidora. Tiene diferencias en su glucosilación postranscripcional lo que disminuye su vida media. Posee un buen perfil de seguridad, informándose un caso de anafilaxia en una paciente con alergia al conejo.22,23 En los estudios realizados se ha demostrado su efectividad en el control de los síntomas del angioedema. Incluso el primer estudio tuvo que detenerse por demostrar ventaja significativa en el tiempo de inicio de la mejoría con su uso comparado con placebo.22,24
Nuevos tratamientos
Ecallantide: La comprensión del mecanismo por el que se desarrolla el angioedema ha sido central para el desarrollo de tratamientos más efectivos. Al identificarse a la bradicinina como principal mediador involucrado en la génesis del angioedema se han desarrollado estrategias para evitar su acción. Ecallantide es un inhibidor específico de la calicreína plasmática producida en la levadura Pichia pastoris.26,27 Se ha aprobado su uso en el 2009 para el episodio agudo en pacientes mayores de 16 años. Se aplica de manera SC a una dosis de 30 mg, alcanzando niveles máximos a las dos o tres horas después de su aplicación SC, con una vida media de dos horas.26,28
Se presentan efectos adversos entre los que destaca la anafilaxia en 4% de los pacientes por lo que se recomienda su uso hospitalario estricto. Dos estudios ADCCP han evaluado su seguridad y eficacia en el tratamiento de los episodios agudos, encontrando mejoría de los síntomas en menor tiempo comparado con el placebo y manteniéndose a las 24 horas. No hubo diferencias con respecto a la localización del angioedema.26
Icatibant: Otro abordaje en el tratamiento del AE esta enfocado a inhibir la capacidad de la bradicinina para unirse con su receptor B2.26 Icatibant es un antagonista competitivo del receptor B2 de la bradicinina de utilidad en los episodios agudos. Se administra SC como una dosis única de 30 mg, obteniéndose una concentración máxima a los 30 minutos con una vida media de una a dos horas. Los efectos adversos son principalmente locales, en el sitio de aplicación del medicamento.25
Tratamiento no recomendado
No se cuenta con evidencia para recomendar el uso de antihistamínicos, corticosteroides o epinefrina para el tratamiento de este padecimiento.1
Situaciones especiales
Embarazo: Como se comentaba previamente, los niveles hormonales tienen efecto sobre el curso de la enfermedad, observándose exacerbaciones durante la pubertad, menstruación, embarazo y uso de agentes hormonales.
Se presentan exacerbaciones en la pubertad en 62%, 35% en la menstruación y en el embarazo 38%.2
Durante el embarazo están contraindicados los andrógenos atenuados. Se recomienda el uso de antifibrinolíticos, dado que no se ha demostrado algún efecto teratogénico. También se recomienda el uso de analgesia regional, a diferencia de la general en la que el se requiere de intubación. Se ha utilizado el concentrado del inhibidor sin relacionarse con efectos adversos.1,2
Anticoncepción: Con respecto al uso de anticonceptivos, se prefieren los que contienen únicamente progestágenos ya que los que contienen estrógenos aumentan los episodios agudos en 60% de los casos. Con respecto al uso de DIU se considera una buena alternativa, generalmente bien tolerado.29
Pediatría: En la infancia se ha identificado que el trauma mecánico y las infecciones son los principales factores desencadenantes, aunque el edema laríngeo se presenta con menor frecuencia, reportándose en 0.9% de los episodios agudos. De presentarse significa un riesgo importante ya que por el menor calibre de la vía aérea en los pacientes pediátricos, el riesgo de asfixia es mayor. Con respecto al diagnóstico, se debe considerar que en pacientes menores de un año se debe confirmar el diagnóstico posterior al año de edad por encontrar niveles menores de INH y del complemento comparado con el adulto.30-32
Conclusiones
El AE es una enfermedad hereditaria poco común, pero de importancia por el riesgo que representa para la vida del paciente. Afecta la calidad de vida por la recurrencia de los síntomas. Actualmente se cuentan con diversas opciones terapéuticas para su tratamiento. Si bien, existen aspectos poco estudiados con respecto al AE entre los que se incluyen la determinación de los factores que determinan la severidad de la enfermedad en cada paciente, el papel del receptor B1 de la bradicinina y su uso como posible blanco terapéutico y la posibilidad de nuevos fármacos administrados en el domicilio del paciente.
Correspondencia: Dra. Andrea Aída Velasco Medina.
Dr. Balmis 148, colonia Doctores. México, D.F. 06720.
Correo electrónico:andreavelasco@hotmail.com