




Presentamos el caso de un paciente portador de una mutación en el gen KCNH2 para el síndrome de QT largo, diagnosticado a raíz de muerte súbita de un familiar. El estudio genético para el síndrome del QT largo y otras canalopatías está siendo cada vez más utilizado en la prevención de la muerte súbita, empezando a aparecer en los últimos años las primeras recomendaciones para el manejo de los pacientes portadores de mutaciones sin expresión fenotípica.
We report a carrier of the KCNH2 gene for the long QT syndrome. He was diagnosed after the sudden death of a relative. The long QT syndrome (and other canalopathies) genetic study is being a new weapon for the prevention of sudden death. In last years, clinical guidelines for the management of genetic carriers without phenotypic manifestations are being published.
Apresentamos um caso de um paciente portador de uma maturação no gene KHNC2 para a síndrome de QT largo, diagnosticado à raiz de morte súbita de um familiar. O estudo genético para a síndrome do QT largo e outras canalopatias estão sendo cada vez mais utilizados na prevenção da morte súbita, começando a aparecer, nos últimos anos, as primeiras recomendações para o tratamento dos pacientes portadores de mutações sem expressão fenotípica.
En los últimos años se han relacionado diferentes mutaciones genéticas con el síndrome del QT largo (SQTL) y dichas mutaciones pueden hallarse en pacientes asintomáticos, sin representación clínica ni electrocardiográfica de la enfermedad1.
El estudio genético de patologías cardíacas potencialmente graves, tiene una especial repercusión en medicina del deporte, donde puede llegar a ser una herramienta muy importante, para el cribado cardiovascular, en un futuro no tan lejano. Presentamos el caso de un paciente diagnosticado de mutación del gen KCNH2.
Caso clínicoVarón de 40 años de edad, deportista habitual (carrera continua 3 horas semanales), fumador de un paquete/día, sin hábito enólico ni otros antecedentes patológicos referidos inicialmente. Consultó en servicio de Cardiología Deportiva para asesoramiento sobre la práctica deportiva, tras identificarse como portador de mutación del gen KCNH2, asociada a SQTL, dentro del estudio familiar de muerte súbita acaecida en un familiar de segundo grado (primo paterno) de 35 años de edad. Aportaba informe de estudio genético que mostraba mutación en el exón KCNH2-8. Dentro de sus familiares directos, su padre también resultó portador de la mutación genética. Dado que se trataba de un estudio familiar sólo se había estudiado la mutación presente en el caso índice (no conocida premortem).
El paciente nunca había sido sometido a ningún estudio de aptitud para el deporte, ni realizaba controles médicos periódicos de otra naturaleza. En la anamnesis dirigida el paciente refirió antecedente de 4 síncopes bruscos, sin pródromos ni relación con el estrés ni el deporte, siendo el último episodio 10 años antes. Los cuadros habían sido atribuidos a episodios vasovagales y sólo se habían realizado electrocardiogramas (ECG) en cada episodio que se habían informado como normales. Nunca presentó episodios relacionados con la práctica deportiva.
En la exploración física el paciente presentaba una auscultación cardíaca sin ruidos patológicos, auscultación respiratoria normal, pulsos periféricos presentes y simétricos, sin ingurgitación yugular ni estigmas de enfermedad de Marfan.
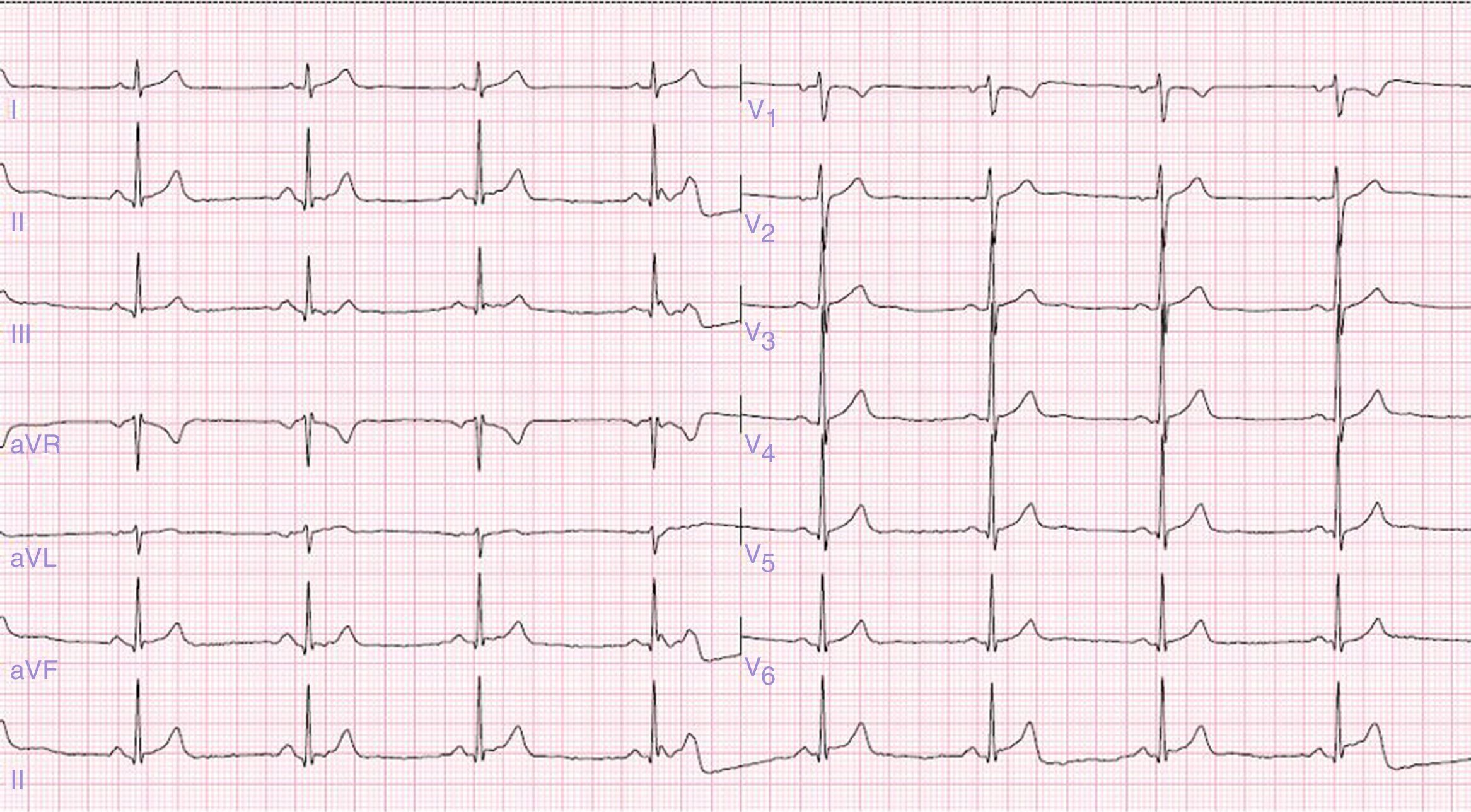
El ECG basal (fig. 1) mostró un ritmo sinusal, a 55 latidos por minuto, con una duración del intervalo QT (QT) de 394ms y un QT corregido por frecuencia cardíaca (QTc) de 366ms.

El paciente computó 1.5 puntos en los criterios diagnósticos de SQTL (tabla 1: síncopes sin estrés 1 punto, frecuencia cardíaca basal 0.5 puntos). Pese a tratarse de un paciente de bajo riesgo, al ser portador del gen KCNH2 se solicitó estudio Holter electrocardiográfico, que mostró tendencia a la bradicardia sinusal sin alteraciones del ritmo y con QT y QTc estables, y ergometría en tapiz rodante (protocolo de Bruce modificado) que resultó normal (progresión del QT y QTc sin anomalías).
Puntuación de Schwartz para el diagnóstico del síndrome del QT largo (1993)
| Hallazgos en ECG | |
|---|---|
| QTc ms (Calculado mediante la fórmula de Bazzet: QTc=QT/√RR) | |
| >480 | 3 |
| 460-470 | 2 |
| 450 (varones) | 1 |
| Torsade de pointes | 2 |
| Alternancia de la onda T | 1 |
| Muescas onda T en 3 derivaciones | 1 |
| Frecuencia cardiaca en reposo por debajo del segundo percentil para la edad | 0.5 |
| Historia clínica | |
| Síncopes con estrés | 2 |
| Síncopes sin estrés | 1 |
| Sordera congénita | 0.5 |
| Historia familiar (el mismo familiar no debe considerarse en ambos) | |
| Familiares de primer grado con SQTL confirmado (criterios de Schwartz puntuación ≥4) | 1 |
| Familiares de primer grado con muerte súbita inexplicada antes de los 30 años | 0.5 |
| Puntuación Schwartz: ≥4 puntos: probabilidad alta; 2-3 puntos: probabilidad intermedia; ≤1 punto: probabilidad baja | |
Modificado de: Schwartz et al.2.
Debido a que el paciente presentaba una bradicardia sinusal se decidió no iniciar tratamiento betabloqueante. Se recomendó al paciente seguir con su actividad deportiva habitual, sin aumentar la intensidad ni la frecuencia, sin supervisión médica. Se explicaron signos de alarma estrictos y se facilitó al paciente un listado con medicamentos susceptibles de alargar el QT (tabla 2) y se le emplazó a realizar un control electrocardiográfico semestral.
Fármacos que prolongan el intervalo QT
| Fármacos antiarrítmicos |
| Quinidina |
| Procainamida |
| Amiodarona |
| Sotalol |
| Antimicrobianos |
| Macrólidos |
| Levofloxacino |
| Moxifloxacino |
| Pentamidina |
| Antihistamínicos |
| Psicotropos |
| Fenotiazinas |
| Tricíclicos y tetraciclicos |
| Haloperidol |
| Metadona |
| Otros medicamentos y sustancias |
| Cisapride |
| Domperidona |
| Cocaína |
| Insecticidas |
Modificado de: Celaya y Martínez3.
El SQTL congénito es una canalopatía hereditaria que afecta aproximadamente a una de cada 2500 personas, principalmente gente joven. Se caracteriza por una prolongación de la repolarización ventricular que se manifiesta por un incremento del QT en el ECG1. Esto expone a los pacientes a presentar síncope y muerte súbita por Torsade de pointes, que degenera en fibrilación ventricular. Hasta la fecha, se han identificado más de 700 mutaciones en 13 genes relacionados con el SQTL de las cuales aproximadamente el 75% se encuentran en 3 genes: KCNQ1 (SQTL tipo 1) y KCNH2 (SQTL tipo 2), que afectan a los canales de potasio, y SCN5A (SQTL tipo 3), que codifica los canales del sodio4,5.
El gen KCNH2 se localiza en el brazo largo del cromosoma 7 (7q36.1) y codifica una subunidad de poro del canal del potasio. La mutación de dicho gen resulta en alteraciones de la repolarización cardíaca, pudiendo afectar al QT. El SQTL es su manifestación más frecuente y potencialmente más grave, pero también se ha relacionado con el síndrome del QT corto y con mayor predisposición al alargamiento adquirido del QT (generalmente farmacológico)5,6.
Pese a que el estudio genético no se encuentra entre los criterios diagnósticos del SQTL5 (tabla 1), diversos estudios han demostrado que tiene una alta sensibilidad para el diagnóstico de la patología y que la mutación más frecuente es la del gen KCNH2. El interés del estudio genético es detectar pacientes portadores de la mutación, con poca expresión fenotípica (exploraciones complementarias normales, principalmente ECG)1,6.
La muerte súbita en sujetos con corazón estructuralmente normal tiene un origen arrítmico en la mayoría de los casos, siendo los trastornos de los canales iónicos la causa más común. De estas, las más prevalentes incluyen el SQTL, el síndrome de Brugada (SB), la taquicardia ventricular catecolaminérgica polimórfica y la fibrilación ventricular idiopática6. Debemos considerar que los portadores silentes de mutaciones genéticas presentan un aumento del 10% riesgo de evento cardíaco entre el nacimiento y los 40 años de edad6.
Pruebas diagnósticasLas pruebas diagnósticas, además de la anamnesis y la exploración física, permiten estratificar el riesgo. Igualmente, en los próximos años, el estudio genético podrá resultar una herramienta clave para el diagnóstico precoz y la prevención de canalopatías y cardiopatías congénitas7.
Ergometría: en condiciones normales el QT se acorta con el ejercicio. En pacientes con SQTL el QT puede alargarse con el esfuerzo o mantenerse estable. La ergometría nos permite diferenciar diferentes tipos de SQTL:
- -
SQTL 1: la respuesta cronotrópica está alterada (no suelen llegar a la frecuencia cardiaca máxima) y el QT se alarga con el ejercicio, incluso puede acompañarse de un aumento de este alargamiento durante la recuperación.
- -
SQTL 2: suelen alcanzar la frecuencia cardiaca máxima y no prolongar o prolongar mínimamente el QT durante el ejercicio. En algunos estudios se ha descrito que prolongan el QT, a los 2 minutos de la fase de recuperación8.
- -
SQTL 3: suelen presentar una respuesta normal del QT con el ejercicio.
Estos distintos comportamientos explican por qué en el SQTL tipo 1 la mayor parte de los eventos cardiacos ocurren durante el ejercicio y en el SQTL tipo 3, durante el reposo. Igualmente, la ergometría nos puede permitir diagnosticar arritmias durante el ejercicio.
Es importante resaltar que la valoración del QT debe incluir el valor del QTc. Este intervalo debe ser determinado, al inicio y final de cada etapa de la ergometría, así como durante la recuperación. Debido a la gran variabilidad interobservador, se recomienda que la lectura de la misma se realice por personal experto9.
La ergometría también puede ser útil para valorar la respuesta al tratamiento y estratificar el riesgo en los casos asintomáticos o que presentan dudas acerca de los factores precipitantes de las arritmias.
Holter: permite una valoración amplia y dinámica del intervalo QT; en ocasiones pueden registrarse episodios espontáneos de arritmia ventricular asintomática, así como posibles episodios de disfunción del nódulo sinusal o bloqueo AV.
TratamientoEn las últimas guías europeas ya se incluyen los betabloqueantes como tratamiento preventivo tanto en pacientes con SQTL, diagnosticado por criterios clínicos o electrocardiográficos (recomendación Ib), como en pacientes portadores asintomáticos del gen (recomendación IIa). En el caso de pacientes con bradicardia sinusal, el tratamiento betabloqueante se puede obviar, siempre que se realice un control clínico estricto10.
ConclusionesEl SQTL es una de las posibles causas de muerte súbita en el deportista. El manejo en el paciente sintomático o con expresión electrocardiográfica es conocido pero en los últimos años se han descrito mutaciones genéticas, que predisponen a padecer dicha patología, aún sin manifestación fenotípica. La aplicación cada vez más extendida del estudio genético, como cribado de patologías cardiovasculares potencialmente graves, está permitiendo descubrir muchos pacientes portadores de este tipo de mutaciones, apareciendo guías de práctica clínica para realizar un abordaje óptimo y disminuir la mortalidad de origen cardiovascular.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
