


La histiocitosis de células de Langerhans es una enfermedad que afecta predominantemente a niños, aunque puede diagnosticarse en la edad adulta. En adultos, la infiltración histiocitaria afecta predominantemente a hueso, pulmón y piel, y presenta especial predilección por el eje hipotálamo-hipofisario. Presentamos el caso de un varón de 51 años que inicialmente solo presentaba diabetes insípida central pero que con el paso del tiempo ha desarrollado una enfermedad sistémica con afectación cutánea, pulmonar, ósea, adenohipofisaria y del sistema nervioso central. La respuesta al tratamiento quimioterápico y radioterápico fue excelente y actualmente no se ha observado progresión de la enfermedad.
Langerhans cell histiocytosis is considered a paediatric disease, although it may be diagnosed in adults. The histiocyte infiltration in adults is most frequently in bones, lungs and skin, and shows a particular predilection for hypothalamus-pituitary axis. A case is presented of a 51 year-old man who initially only presented with central insipidus diabetes, and over time developed systemic disease with skin, lungs, bones, pituitary, and central nervous system involvement. Chemotherapy and radiotherapy were effective, and there is currently no progression of the disease.
La histiocitosis de células de Langerhans (LCH) es una enfermedad granulomatosa de etiología desconocida1. La prevalencia es de 3-4 casos por millón en niños y significativamente menor en adultos2. En los adultos, la infiltración histiocitaria afecta frecuentemente a hueso, pulmón y piel, y presenta especial predilección por el eje hipotálamo-hipofisario con diabetes insípida hasta en el 50% de los casos3.
Describimos el caso de un paciente adulto que consultó por clínica compatible con diabetes insípida central y resultó ser secundaria a LCH.
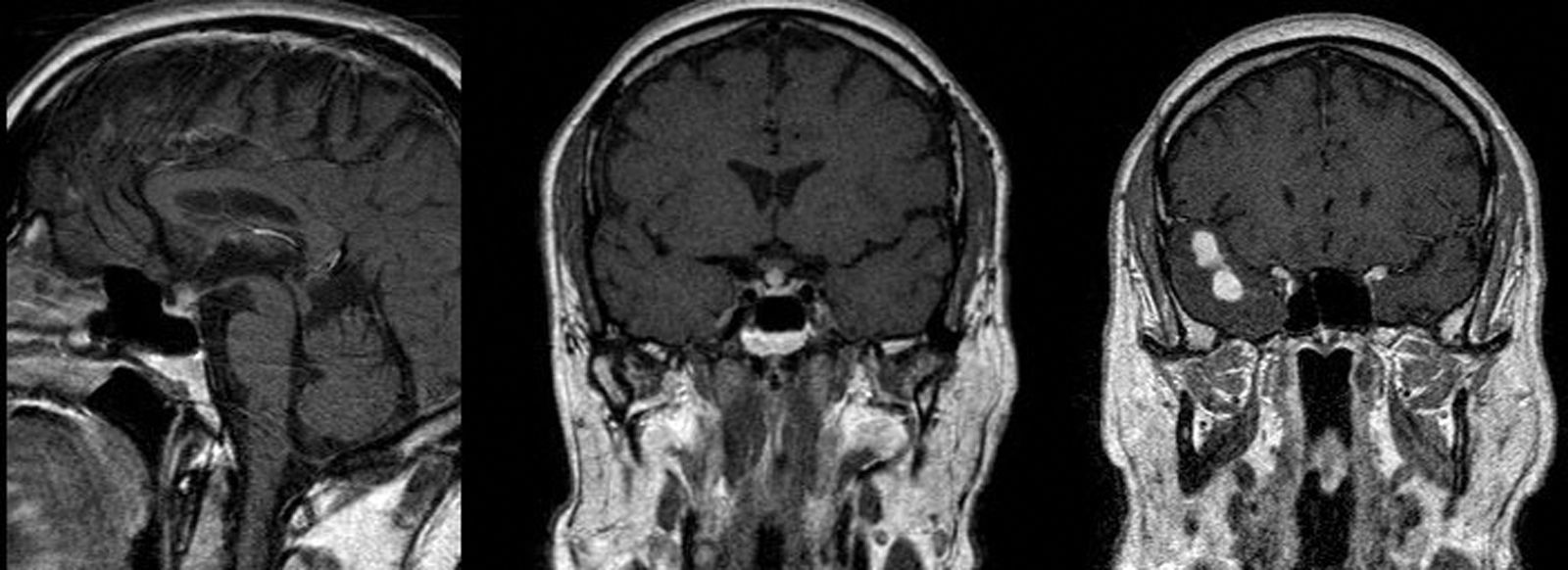
Caso clínicoVarón de 51 años, exfumador y diagnosticado de EPOC, que consultó por poliuria y polidipsia intensa con nicturia de inicio brusco de un mes de evolución. No refería cefaleas ni otra sintomatología acompañante. Se cuantificó una diuresis de 15 l/día con osmolaridad baja e hipernatremia con osmolaridad elevada en plasma, por lo que no se realizó un test de deshidratación. Con desmopresina oral presentó clara mejoría de los síntomas. La RM de silla turca demostró una hipófisis aumentada de tamaño sin imagen de tumor delimitable, con el tallo engrosado y ausencia de señal hiperintensa en T1 de la neurohipófisis, indicativo de hipofisitis (fig. 1). La función adenohipofisaria y la campimetría fueron normales. En ese momento se desestimó la realización de una biopsia hipofisaria.

A los 4 meses se objetivó probable déficit de hormona de crecimiento con factor de crecimiento insulínico tipo 1 44,7ng/ml (normal para su edad y sexo 92-245ng/ml) e hipogonadismo hipogonadotrófico: hormona folículo estimulante 0,5 U/l (1,7-11), hormona luteinizante 0,2 U/l (0,5-6) y testosterona total 0,17ng/ml (2,4-10,7), que se trató con undecanoato de testosterona trimestral.
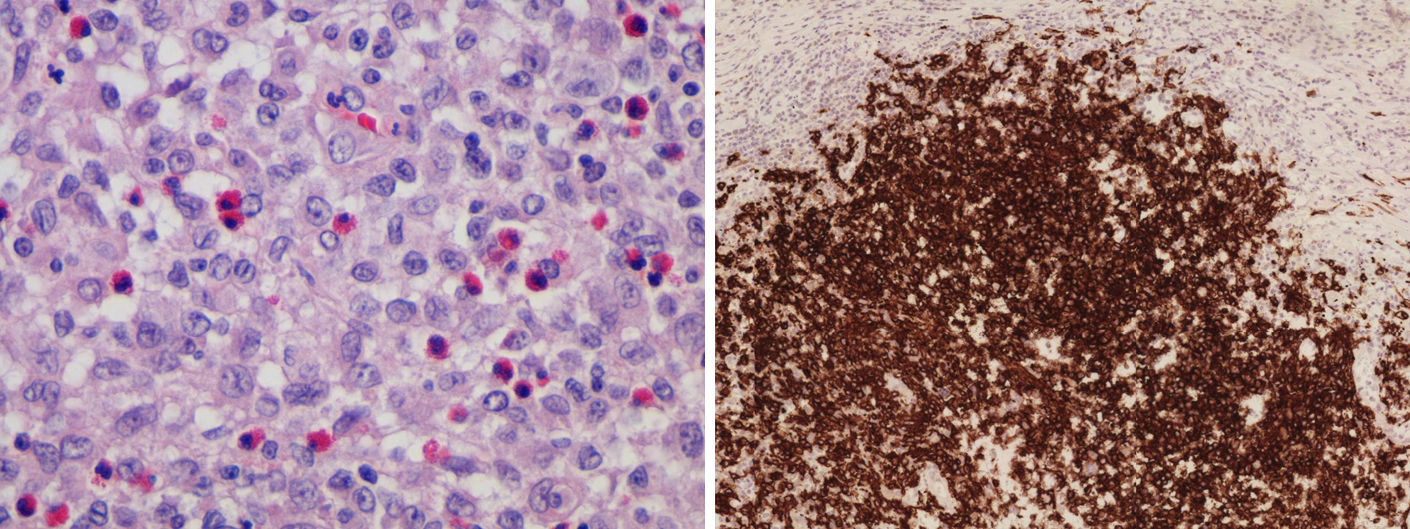
Al año del diagnóstico, en la RM de control se observó una disminución de tamaño de la hipófisis y del tallo, pero en la región temporal derecha se observaban 2 nódulos subcorticales de 10 y 15mm con edema perilesional que no se apreciaban en la RM previa (fig. 1). El paciente fue intervenido por el servicio de Neurocirugía y el estudio anatomopatológico de dichas lesiones confirmó la proliferación de histiocitos de Langerhans con inmunohistoquímica positiva para CD1a, S100 y CD68, todo ello compatible con una LCH (fig. 2).

Evaluado por los servicios de Hematología, Dermatología y Neumología, se confirmó la afectación cutánea y pulmonar, y se descartó infiltración ósea, ganglionar y de médula ósea. Se administró un único ciclo de vinblastina, mercaptopurina y prednisona, con buena tolerancia al tratamiento.
A los 2 años, consultó por dolor en la cadera y se observó afectación ósea a nivel de hueso ilíaco, que se trató con radioterapia, con buena respuesta antiálgica.
Actualmente, a los 5 años del diagnóstico, el paciente se encuentra asintomático, no ha habido progresión de la enfermedad y sigue tratamiento con desmopresina y testosterona. Mantiene intactos los ejes hipotálamo-hipófiso-tiroideo y adrenal.
DiscusiónLa LCH es una enfermedad granulomatosa caracterizada por la proliferación de células dendríticas específicas, las células de Langerhans, que derivan del sistema macrófago-monocítico1. Dichas células pueden infiltrar cualquier tejido u órgano sin producir disfunción necesariamente. La edad de presentación más habitual es la infancia, entre 1-3 años, aunque puede manifestarse a cualquier edad2. En adultos la edad media al diagnóstico es de 33 años, con predominio del sexo masculino (2:1)2. El diagnóstico requiere la demostración de gránulos de Birbeck mediante microscopia electrónica y la presencia de CD1o langerina en la superficie celular4.
El curso de la enfermedad es impredecible, pudiendo resolverse espontáneamente o manifestarse como enfermedad diseminada3. Según la edad, se observan 2 fenotipos distintos; en adultos afecta a pulmón, hueso, piel y región hipotálamo hipofisaria, mientras que en los niños a hígado, bazo, ganglios linfáticos y médula ósea5.
La diabetes insípida es la manifestación endocrina más frecuente3-6. Dependiendo de las series publicadas, la prevalencia puede llegar hasta el 50% si se incluyen pacientes con enfermedad sistémica3-7. Aunque se suele desarrollar durante el primer año del diagnóstico puede preceder a este8, como ocurrió en nuestro caso, o bien desarrollarse a lo largo del curso de la enfermedad7. La diabetes insípida habitualmente es irreversible5,7. Los hallazgos radiológicos en la RM, engrosamiento del tallo hipofisario con ausencia de la mancha blanca neurohipofisaria, hacen difícil el diagnóstico diferencial entre germinoma, neurosarcoidosis, hipofisitis, tuberculosis e incluso diabetes insípida idiopática6,9. Si hay afectación adenohipofisaria, la hormona de crecimiento es la primera en afectarse seguida de las gonadotrofinas y la tirotrofina5,7. El déficit glucocorticoide se ha descrito tan solo en el 1-2%, la mayoría en contexto de panhipopituitarismo5,7. Existen otras manifestaciones endocrinas descritas, como la infiltración tiroidea, paratiroidea, adrenal o pancreática, siempre en pacientes con enfermedad diseminada5.
Actualmente existe una guía para el manejo de la LCH en niños y recientemente se han publicado unas recomendaciones realizadas por un grupo de expertos para adultos4. Si existe afectación hipotálamo hipofisaria, el tratamiento hormonal sustitutivo será el estándar, dependiendo del déficit encontrado. No hay estudios sobre el tratamiento con hormona de crecimiento en adultos. Si hay afectación ósea, recomiendan tratamiento con corticoides intralesionales o radioterapia. En cambio, si la enfermedad está diseminada, tratamiento quimioterápico con vinblastina y glucocorticoides, como se hizo en nuestro caso.
En conclusión, la LCH debe considerarse en el diagnóstico diferencial de pacientes con diabetes insípida central, déficit anterohipofisario y patología selar. Aunque inicialmente no tengan alteración hormonal, lo pueden desarrollar en el curso de la enfermedad, por lo que la monitorización por parte del endocrinólogo debe ser obligatoria. Dada la posible afectación de diferentes órganos o tejidos, es fundamental el seguimiento multidisciplinario de estos pacientes para el correcto manejo y tratamiento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.