




Monkeypox (Mpox) is a zoonotic disease caused by the monkeypox virus (MPXV). MPXV can be transmitted by close contact with lesions, body fluids, respiratory droplets, and contaminated materials. A new pattern of spread among sexual networks has been recently described. The present work aimed to report the epidemiological and genomic characterization of the 2022 MPXV outbreak in central Argentina. A total of 113 scabs and/or lesion swab specimens were studied. MPXV infection was confirmed in 46.0% of the studied patients, all of whom were men. Varicella-zoster virus infection was the most frequent differential diagnosis. Eight complete viral genomes were obtained by next-generation sequencing. The Argentinian sequences were grouped intermingled with other sequences from the 2022 MPXV outbreak, related to samples from the USA, Europe, and Peru. Taken together, our study provided an initial assessment of the genetic and epidemiological characteristics of the 2022 MPXV outbreak in Córdoba, Argentina.
La viruela del mono es una enfermedad zoonótica causada por el virus de la viruela del mono (MPXV). El MPXV puede transmitirse por contacto con lesiones, fluidos corporales, gotitas respiratorias y materiales contaminados. Recientemente, se ha descrito un nuevo patrón sexual de transmisión. El presente trabajo tuvo como objetivo caracterizar epidemiológica y molecularmente el brote de MPXV de 2022 en la región central de Argentina. Se estudiaron 113 muestras de costras y/o hisopados de lesiones. La infección por MPXV se confirmó en el 46,0% de los casos, todos ellos hombres. La infección por el virus varicela-zoster fue el diagnóstico diferencial más frecuente. Se obtuvieron ocho genomas completos mediante la secuenciación de última generación. Las secuencias argentinas se agruparon entremezcladas con otras secuencias del brote de MPXV de 2022, relacionadas con las de EE. UU., Europa y Perú. Nuestro estudio proporcionó información sobre las características del brote de MPXV de 2022 en Córdoba, Argentina.
Mpox is a zoonotic disease caused by the monkeypox virus (MPXV), a member of the Orthopoxvirus genus and close relative of the variola virus (smallpox virus)8. It has a double-stranded DNA genome consisting of ∼197kb with ∼190 non-overlapping coding genes, which are responsible for its various biological characteristics (immunomodulation, host preference/diversity, and viral pathogenicity)10.
The virus can be transmitted from human to human by close contact with lesions, body fluids, respiratory droplets, and contaminated materials8. A new pattern of spread among sexual networks, specifically among men who have sex with men (MSM) has been recently described and underscored sexual contact as a main spreading route for the virus among other potential sources5.
Historically, MPXV has been classified into two main clades: West African and Central African. However, a new proposal for classifying MPXV has been established in order to group isolates into three clades7, each with distinct geographical, clinical, genomic, and epidemiological differences1.
The first two clades include most isolates linked to outbreaks from the Democratic Republic of Congo (MPXV Clade 1), formerly known as the Congo Basin Lineage, and from West Africa (MPXV Clade 2), known as the West Africa Lineage. Isolates from these clades, which are responsible for most of the natural transmission cycles and endemism in Africa, reveal different transmission, pathobiological patterns, and distinct clinical outcomes that further demonstrate differentiation among them14. Clade 1 is more clinically severe, with higher mortality rates, and exhibits increased transmissibility. Conversely, Clade 2 MPXV isolates are associated with milder infection, lower mortality rates, and reduced transmissibility9. Interestingly, Clade 3 (MPXV Clade 3) includes isolates from the 2017 to 2019 outbreaks and genomes from the most recent 2022 outbreak divergent emerging lineages7. On May 7, 2022, the WHO was informed of a confirmed case of mpox in an individual who traveled from the United Kingdom to Nigeria and subsequently returned to the United Kingdom and represented the index case of this outbreak15.
In Argentina, on May 22, 2022, a notification of a patient with suspected monkeypox virus infection was received. It corresponded to an individual from the province of Buenos Aires, who consulted for itching, pustules in different parts of the body, fever, and lower back pain, symptoms that began on May 15, 2022. The patient had a history of travel to Spain between April 28 and May 16, 2022. The case was confirmed by the Argentine Ministry of Health on May 27, 202212.
With regard to the province of Córdoba, a central area of the country, the first case of mpox was reported on June 25, 2022, in a male patient who, upon returning from a trip to Mexico, presented the characteristic skin lesions13.
The present work aimed to report the identification and sequencing of the complete genome of MPXV in clinical samples from patients treated in the health system of the province of Córdoba, and to describe the clinical–epidemiological characteristics of these patients and the differential diagnosis of the disease.
A total of 113 skin lesion samples (scabs and/or lesion swab specimens) from patients with suspected MPXV infection who had access to public health centers of Cordoba (the second most populated inland province of Argentina), were studied between July 2022 and March 2023. Cordoba city is the capital of the province of Cordoba. It is located in the central region of Argentina (31°25′00″S 64°11′00″O) and has 1505250 inhabitants with a population density of 2308inhabitants/km2 (INDEC, 2022)2.
Samples were subjected to DNA extraction using the MagNA Pure 96 DNA and Viral NA Large Volume Kit (Roche, Germany). Subsequently, a real-time PCR (qPCR) using the TaqMan™ Monkeypox Virus Microbiome Detection Assay (Applied Biosystems, USA), designed for the qualitative detection of MPXV from the West African and Congo-Basin clades was performed. Positive samples were subjected to NGS sequencing.
A high throughput shotgun metagenomics strategy was implemented to obtain the complete sequence genome of the virus. For that purpose, a library preparation was performed using an adapted protocol with the COVIDseq kit (Illumina, USA), utilizing the library preparation reagents, starting from the tagmentation step. The sequencing was carried out on the MiSeq platform (Illumina, USA) by paired-end sequencing (2×150 nucleotides) and an average fragment size of 350 nucleotides.
Illumina reads were mapped to the MPXV genome (GenBank NC_063383.1 – Clade 3/Lineage A.1) using BWA-MEM v.0.7.1711 and a consensus sequence was obtained by piping a SAMtools v.1.74 pileup with iVar v.1.3.13 consensus as described elsewhere.
Initially, clade and lineage assignment were performed using Nextclade v2.5.0 tool (https://clades.nextstrain.org/). Subsequently, a phylogenetic analysis was conducted using the maximum likelihood method, under the appropriate nucleotide substitution model (TVM+G) selected by jModeltest, according to the Akaike Information Criterion. The robustness of the reconstructed phylogeny was evaluated by the bootstrap analysis (10000 replicates) using the IQ-Tree program (http://iqtree.cibiv.univie.ac.at/).
The study was carried out on the same biological sample sent for the diagnosis of MPXV infection. The project was approved by the institutional Training and Teaching Committee and the information confidentiality was guaranteed.
Nucleotide sequences obtained in this work were deposited in the GISAID database under accession numbers: EPI_ISL_15972402 to EPI_ISL_15972409.
The mean age of the population analyzed was 35.6 (±8.5) years and 88.5% (100/113) were male. MPXV infection was confirmed in 46.0% (52/113) of the studied patients, all of whom were men. Among the positive cases, 57.7% (30/52) were HIV (+).
The most prevalent symptoms reported were: fever [61.5% (32/52)] followed by asthenia [46.2% (24/52)] and lymphadenopathy [42.3% (22/52)]. Myalgia was present in 38.5% (20/52) of the cases, and the least commonly reported symptoms were low back and anorectal pain. With regard to skin symptoms, the most frequent lesions were vesicular rash and pustules [40.4% (21/52) each], followed by scabs [38.5% (20/52)], umbilicated lesions and maculopapular rash [30.7% (16/52) each].
Seven cases (13.5%) had a history of travel to countries/cities where cases of mpox were recorded (Spain, Mexico, Paraguay, Brazil, and Buenos Aires city), being the first cases reported in our city, while subsequent cases [86.5% (45/52)] were related to the local circulation of the virus.
Concerning sexual orientation, 44.2% (23/52) reported having same-sex sexual partners, 3.8% (2/52) were men having sex with women, and 51.9% (27/52) data were not recorded. Despite this fact, sexual exposure could be documented in 80.8% (42/52) of cases, most of which reported genital and/or (peri-)anal lesions. On the other hand, some risk factors were identified such as multiple/occasional sexual partners [50% (26/52)], unprotected sexual intercourse [73.1% (38/52)], and having sex with a confirmed case or travelers from countries with confirmed cases [28.8% (15/52)].
Finally, a differential diagnosis was obtained in 12 patients with negative results for MPXV. Of these, 75.0% (9/12) corresponded to varicella-zoster virus infections, 16.7% (2/12) to herpes simplex virus type 2 infection and 8.3% (1/12) to acute retroviral syndrome.
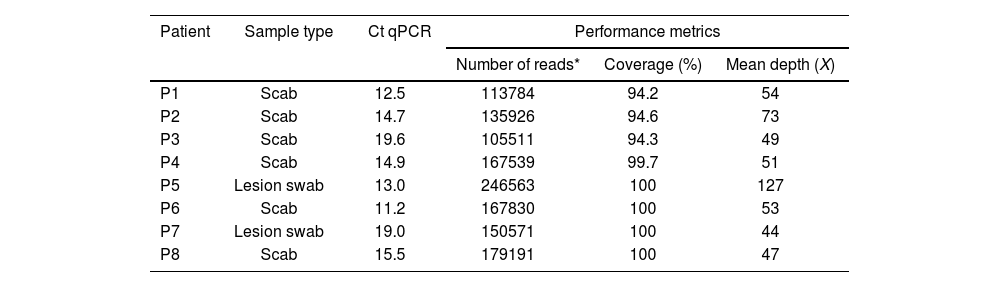
The NGS protocol was subjected to 14 positive samples (oropharyngeal swabs, lesion swabs, or scab samples – qPCR Ct <29.5), obtaining 8 MPXV complete genomes. Five million total reads were obtained per sample, with a genome-wide coverage depth of 44× to 127× (mean: 62×). The genome coverage obtained was 94.2% to 100% (mean: 97.9%) (Table 1).
NGS protocol performance metrics.
| Patient | Sample type | Ct qPCR | Performance metrics | ||
|---|---|---|---|---|---|
| Number of reads* | Coverage (%) | Mean depth (X) | |||
| P1 | Scab | 12.5 | 113784 | 94.2 | 54 |
| P2 | Scab | 14.7 | 135926 | 94.6 | 73 |
| P3 | Scab | 19.6 | 105511 | 94.3 | 49 |
| P4 | Scab | 14.9 | 167539 | 99.7 | 51 |
| P5 | Lesion swab | 13.0 | 246563 | 100 | 127 |
| P6 | Scab | 11.2 | 167830 | 100 | 53 |
| P7 | Lesion swab | 19.0 | 150571 | 100 | 44 |
| P8 | Scab | 15.5 | 179191 | 100 | 47 |
The phylogenetic analysis revealed that all MPXV strains sequenced during this study clustered closely related to each other, suggesting that the 2022 outbreak would have a single origin. The 2022 sequence group (B.1 lineage) forms a descended divergent branch from a branch with MPXV strains belonging to A.1 lineage associated with MPXV export from Nigeria (outbreak in 2017–2018) to several countries (United Kingdom, Israel, and Singapore) in 2018 and 2019 (Fig. 1). The newly detected MPVX strains clustered together inside a highly supported clade containing West Africa (Clade 3/B.1 lineage), and MPVX sequences recently detected in Europe and the USA (Fig. 1).
 (http://iqtree.cibiv.univie.ac.at/).")
Maximum likelihood phylogenetic tree constructed using MPXV sequences obtained from crust samples from individuals with confirmed MPXV infection from Córdoba, Argentina, and reference sequences from each clade available in GenBank. The phylogenetic tree was constructed with IQ-TREE web server (ultrafast bootstrap values: 10000 replicates) (http://iqtree.cibiv.univie.ac.at/).
The Argentinian sequences grouped intermingled with the other sequences from the 2022 MPXV outbreak, related to samples from the USA, Europe, and Peru. Sequences P2, P3, and P4 obtained from samples from patients with a history of travel to Spain, and close contact with a confirmed case, grouped closely together, forming a highly supported separate clade (Fig. 1).
Studied cases of the MPXV last outbreak suggest that the virus may have made its way into highly interconnected sexual networks within the MSM community (who change partners more frequently and are more likely to have multiple partners at the same time), spreading in ways that differ from those observed in the general population. In a densely connected network, the virus is less likely to reach a dead end.
A new pattern of spread among sexual networks, specifically among men who have sex with men (MSM), has underscored sexual contact as a main spreading route for the virus5. This suggests sexual contact as the main transmission route, unlike previous mpox outbreaks, where the most frequent routes of transmission were respiratory or skin-to-skin contact.
The differential diagnosis of MPXV infection can be complex, including infectious and non-infectious diseases. It is necessary to consider the type of lesions and their location, and the person's immune status6. Nevertheless, in our study a differential diagnosis could be obtained in 19.7% of the negative cases, mainly detecting the varicella-zoster virus.
It was possible to sequence the first MPXV genomes from Argentina, using NGS technology (Illumina). The implemented protocol allowed the sequence of 8/14 (57.1%) complete genomes in samples with a Ct value less than 20, being a successful tool for the study of this virus and adding information to the global monitoring of MPXV variants, providing knowledge about viral dissemination into the population.
All MPXV strains sequenced clustered closely with each other (Clade 3/B.1 lineage), suggesting that the 2022 outbreak would have a single origin. However, it is still unclear if the MPXV-2022 outbreak strain originated from humans or other hosts.
Advances in the development of NGS kits during the COVID-19 pandemic made it possible to have tools that could be adapted for the sequencing of other genomes, such as MPXV. Taken together, our study provided a preliminary assessment of the genetic and epidemiological characteristics of the 2022 MPXV outbreak in Córdoba, Argentina.
FundingThis research has not received any specific grant from agencies in the public, commercial, or non-profit sector.
Conflicts of interestThe authors declare no conflict of interest.
We sincerely thank Dra. Viviana Ré and Dra. Belén Pisano for taking the time to review our manuscript.
