






Escherichia coli O157:H7 is a foodborne pathogen implicated in numerous outbreaks worldwide that has the ability to cause extra-intestinal complications in humans. The Enteropathogens Division of the Central Public Health Laboratory (CPHL) in Paraguay is working to improve the genomic characterization of Shiga toxin-producing E. coli (STEC) to enhance laboratory-based surveillance and investigation of foodborne disease outbreaks. Whole genome sequencing (WGS) is proposed worldwide to be used in the routine laboratory as a high-resolution tool that allows to have all the results in a single workflow. This study aimed to carry out for the first time, the genomic characterization by WGS of nine STEC O157:H7 strains isolated from human samples in Paraguay. We were able to identify virulence and resistance mechanisms, MLST subtype, and even establish the phylogenetic relationships between isolates. Furthermore, we detected the presence of strains belonging to hypervirulent clade 8 in most of the isolates studied.
Escherichia coli O157:H7 es un patógeno transmitido por alimentos implicado en numerosos brotes en todo el mundo y es capaz de causar complicaciones extraintestinales en humanos. La sección de «Enteropatógenos» del Laboratorio Central de Salud Pública trabaja en mejorar la caracterización genómica de STEC, de modo de potenciar la vigilancia laboratorial y la investigación de brotes de enfermedades transmitidas por alimentos. La secuenciación de genoma completo (WGS, por sus siglas en inglés) se propone a nivel mundial como una herramienta de alta resolución para ser utilizada en el laboratorio de rutina, ya que permite obtener todos los resultados en un único proceso. El objetivo de este trabajo fue llevar a cabo, por primera vez, la caracterización genómica por WGS de nueve cepas STEC O157:H7 aisladas en Paraguay a partir de muestras de origen humano. Pudimos identificar los factores de virulencia, los mecanismos de resistencia, el subtipo MLST, e incluso pudimos establecer la relación filogenética entre los aislamientos. Además, detectamos que la mayoría de las cepas pertenecían al clado hipervirulento 8.
Shiga toxin-producing Escherichia coli (STEC) strains constitute a subset of pathogenic E. coli that have been associated with sporadic cases and outbreaks of diarrhea (D), bloody diarrhea (BD) and hemolytic uremic syndrome (HUS) in humans17–19. STEC strains are characterized by the production of Shiga-toxins (Stx), which are AB5 cytotoxins encoded on the bacterial chromosome that inhibit protein synthesis and can cause cell death3. E. coli O157:H7 is the most well-known Stx-producing serotype within this group and most associated with severe disease and outbreaks25,39,42. There are two types of Stx (Stx1 and Stx2) and each one is associated with a different clinical outcome4,21. In fact, in mouse models, Stx2 has shown to be 100 times more potent than Stx143. Moreover, in primate models the administration of Stx2 alone can produce symptoms of HUS, which are not produced by the administration of Stx1 alone38,40.
Stx1 and Stx2 are further divided into subtypes associated with different levels of virulence37. For instance, Stx2a, Stx2c, Stx2d are more often associated with HUS. In contrast, the other Stx1 and Stx2 subtypes are mainly associated with mild disease4,21. Besides Stx, additional virulence factors contribute to the pathogenicity of the STEC strains associated with severe diseases. The most common ones are located in the pathogenicity island called the locus of enterocyte effacement (LEE) and the O157 plasmid17,30,31. The LEE island encodes several proteins needed to produce attaching and effacing (A/E) lesions on the intestinal mucosa, such as intimin (encoded by the eae gene) that closely attaches the bacteria to the intestinal epithelium, the translocated intimin receptor (Tir), and other Type 3 secretion system (T3SS) effectors17,29. The O157 plasmid encodes for several virulence proteins, i.e., enterohemolysin (ehxA), a catalase-peroxidase (KatP), extracellular serine-protease (espP), adhesines (toxB) and proteins of T3SS27.
STEC O157:H7 populations have shown to be genotypically diverse, and variations of disease severity in humans are associated with specific genotypes. Three main lineages (I, II, and I/II) differently distributed between animals, humans and geographical regions were described22,47. Furthermore, O157:H7 strains can be divided into nine clades based on outbreak studies associated with different clinical outcomes28. Strains belonging to clade 8 are known to be “hypervirulent” because they are associated with more severe disease and the potential capacity to cause severe outbreaks28,36. Although the biological basis behind the increased pathogenicity of these strains is not fully understood, it is suggested that the differential expression of some genes could play this role1. Moreover, Kulasekara et al.23 found 7 coding sequences postulated as putative virulence determinants when comparing the genome of the TW14359 strain (clade 8) against two other sequenced strains from clade 1 and 3 (EDL-933 and Sakai).
Epidemiological surveillance of STEC is a fundamental public health issue to limit the spread, detect outbreaks, and trace back contamination sources, allowing the development of efficient and targeted intervention strategies. In Paraguay, the STEC surveillance program was implemented in 2002. Since then, 40 STEC strains have been isolated from clinical samples, 10 of which were attributed to the O157:H7 serotype (CPHL – Paraguay). This work aimed to achieve the genomic characterization and study the genetic diversity of STEC O157:H7 strains isolated from human samples in Paraguay to identify the subtypes associated with high pathogenicity in order to provide insights into the risk they pose to public health.
Materials and methodsBacterial strainsFrom 2002 to 2018, 10 E. coli O157:H7 strains were detected in stool samples from BD or HUS cases in Paraguay through the STEC surveillance program. For this study, 9 out of 10 strains were retrieved and subjected to the traditional genotypic characterization and genomic analysis by WGS at the CPHL in Paraguay and INEI-ANLIS “Dr. Carlos G. Malbrán” in Argentina.
CharacterizationTraditional characterization was performed by serotyping, biotyping and PCR genotyping of the main virulence factors (stx, eae, exhA) following the protocol described in the Procedure Manual for the Diagnosis and Characterization of Shiga-toxin producing E. coli from human samples by molecular biology techniques15 (INEI/ANLIS Malbrán).
Stx subtyping was performed as described by Scheutz et al.37. The strains were subtyped by PFGE using the XbaI enzyme according to the International PulseNet protocol35. The subsequent analysis was performed using the GelCompar II software version 5.1 (Applied Maths, Sint-Martens-Latem, Belgium) using the Dice coefficient of similarity and the UPGMA (Unweighted Pair Group Method with Arithmetic mean) grouping method to construct dendrograms (tolerance 1.5%) and determine the genetic relationship.
The resistance profile was determined by testing the following antibiotics: ampicillin (AMP), amoxicillin/clavulanic acid (AMC), azithromycin (AZI), cefotaxime (CTX), ceftazidime (CTZ), nalidixic acid (NAL), gentamicin (GEN), ciprofloxacin (CIP), tetracycline (TCY), trimethoprim/sulfamethoxazole (SXT), chloramphenicol (CHL), nitrofurantoin (NIT), by agar diffusion (Kirby-Bauer method) following the CLSI (Clinical and Laboratory Standards Institute) regulations7 (2019 version).
Whole-genome sequencingGenomic DNA (gDNA) was extracted and purified from the isolates using Magpurix equipment with the MagPurix Bacterial DNA Extraction Kit by Zinexts Life Science Corp. DNA concentrations were determined using the QuantiFluor® dsDNA System reagent kit with the Quantus™ Fluorometer (Promega Corporation).
Libraries were prepared using the Nextera XT DNA library prep kit (Illumina) according to the manufacturer's guidelines.
Sequencing was performed by the genomic platform of INEI/ANLIS Malbrán (Buenos Aires – Argentina) and by the CPHL (Asuncion/Paraguay) on the Illumina MiSeq platform using the MiSeq Reagent Kit V2.
SerotypingThe Short Read Sequence Typing performed the serotyping of bacterial pathogens using the (SRST2) (v.2.0) tool14. The reads in FASTQ format were mapped against the reference database (EcOH database available at https://github.com/katholt/srst2) in FASTA format using the following parameters: 85% threshold for sequence identity and 60% minimum length for a hit.
Virulome and AMR genesThe prediction of virulence genes was performed from the short-reads through the ARIBA software13 (Ariba-2.14.4 version) using the virulence finder database16 (2020-05-29 version) (available at https://bitbucket.org/genomicepidemiology/virulencefinder_db/src/master/) with a threshold of 95% identity and a minimum length of 80% for a hit.
The Stx subtype was determined from the assembled reads in FASTA format by an in-silico PCR performed using the Ipcress program developed by Guy St.C. Slater (version 0.8.3) using the primers described by Scheutz et al.37. Additional Stx subtyping was performed with the program E. coli Shiga-toxin-typing v.2.0 (Blast v2.9 against Shiga toxin-type databases from the Statens Serum Institut SSI and the Technical University of Denmark DTU available at https://github.com/aknijn/shigatoxin-galaxy) in the Galaxy platform (Galaxy version 2.0 https://aries.iss.it/).
Putative virulence factors described by Kulasekara et al.23 were screened by in-silico PCR using the Ipcress program for the locus tags ECSP_3620 (encoding the anaerobic nitric oxide reductase NorV), ECSP_0242 (encoding a domain that facilitates protein–protein interactions), ECSP_3286 (encoding a product that binds with high affinity to heme), ECSP_2870/2872 (encoding proteins that are involved in the adaptation to plant hosts), ECSP_2687 (encoding proteins that reduce the host immune response) and ECSP_1773 (encoding a protein that interferes with innate immunity).
Antimicrobial resistance (AMR) was screened from both short-reads and assemblies in FASTA format. The short-reads were mapped against Res_Finder database46 (v.4.1) (available at https://bitbucket.org/genomicepidemiology/resfinder_db/src/master/) using the ARIBA software with requirements of 95% identity threshold and a minimum length of 80%. Additional AMR screenings were performed using the AMRfinder program9 from FASTA files.
Plasmid search was conducted by mapping FASTA files against Plasmid_Finder database6 (available at https://bitbucket.org/genomicepidemiology/plasmidfinder_db/src/master/).
Epidemiological typing and phylogenetic analysisIn-silico multilocus sequence typing (MLST) was performed by comparing the raw reads in FASTQ format with the PubMLST database downloaded from the PubMLST website (https://pubmlst.org) using the ARIBA software. Seven house-keeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) were used for MLST analysis.
The lineage-specific polymorphism assay (LSPA-6) was performed by in-silico PCR using the Ipcress program according to the scheme described by Yang et al.45 Isolates with genotype 111111 were classified as LSPA-6 LI, those with genotype 211111 as LSPA-6 LI/II and those with other derivations were classified as LSPA-6 LII.8
Determination of O157:H7 clades was performed by in-silico PCR using the Ipcress program according to the 4 SNPs algorithm developed by Riordan et al.36.
In addition to the genomic characterization, we decided to study the genomic relatedness among our isolates and other E. coli O157:H7 isolated from humans around the world. To accomplish that goal, we submitted the short-read sequences to EnteroBase48 (v1.1.2). We determined the closest relatives to our population of E. coli isolates, taking the HC100 cluster information obtained by using the hierarchical clustering (HierCC) algorithm in EnteroBase and chose 50 closest relatives worldwide whose FASTQ files were available at SRA GenBank from the same period that our strains were collected. Both phylogenetic analyses (one between our nine E. coli O157:H7 strains and the other between our strains and the 50 selected) were done from the paired end reads that were mapped against the E.coli O157:H7_str_EDL933 genome used as a reference. For this purpose, we used Lyve-SET, a pipeline that uses high-quality single-nucleotide polymorphism (hqSNPs) to create a phylogeny20. The output was then used as an input with the FigTree program developed by Andrew Rambaut (version 1.4.4.) to build the phylogenetic tree. Finally, trees obtained from FigTree were used in the iTOL v6 program26 (https://itol.embl.de) for a friendlier visualization.
Publicly available genomesAll raw sequence data for the Paraguayan E. coli genomes analyzed in this publication are publicly available under Bioproject IDs PRJNA277984 and PRJNA764717. The accession numbers of the Paraguayan and the other strains used in the analyses are given in Supplementary Table 1.
ResultsThe genetic characterization showed that all the strains (9) carried the stx2 gene. Furthermore, the subtypes detected were: stx2c (5/9 strains), stx2a+c (3/9) and stx2a (1/9). All the strains harbored eae, ehxA, rfbO157, and fliCH7 genes. With regard to the antibiogram, 4 strains were ampicillin-resistant. At the same time, one ampicillin-resistant strain was also resistant to trimethoprim–sulfamethoxazole.
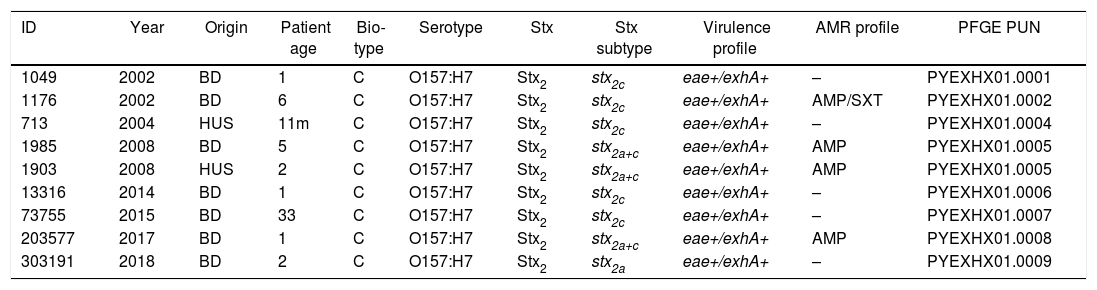
XbaI-PFGE, 7/9 strains generated different patterns except for two strains isolated in the same year and month (1985 and 1903) that showed one unique pattern (PYEXHX01.0005). These results are shown in Table 1, along with the epidemiological information about the isolates.
Traditional characterization results, epidemiological information and PFGE profile of the isolates.
| ID | Year | Origin | Patient age | Bio-type | Serotype | Stx | Stx subtype | Virulence profile | AMR profile | PFGE PUN |
|---|---|---|---|---|---|---|---|---|---|---|
| 1049 | 2002 | BD | 1 | C | O157:H7 | Stx2 | stx2c | eae+/exhA+ | – | PYEXHX01.0001 |
| 1176 | 2002 | BD | 6 | C | O157:H7 | Stx2 | stx2c | eae+/exhA+ | AMP/SXT | PYEXHX01.0002 |
| 713 | 2004 | HUS | 11m | C | O157:H7 | Stx2 | stx2c | eae+/exhA+ | – | PYEXHX01.0004 |
| 1985 | 2008 | BD | 5 | C | O157:H7 | Stx2 | stx2a+c | eae+/exhA+ | AMP | PYEXHX01.0005 |
| 1903 | 2008 | HUS | 2 | C | O157:H7 | Stx2 | stx2a+c | eae+/exhA+ | AMP | PYEXHX01.0005 |
| 13316 | 2014 | BD | 1 | C | O157:H7 | Stx2 | stx2c | eae+/exhA+ | – | PYEXHX01.0006 |
| 73755 | 2015 | BD | 33 | C | O157:H7 | Stx2 | stx2c | eae+/exhA+ | – | PYEXHX01.0007 |
| 203577 | 2017 | BD | 1 | C | O157:H7 | Stx2 | stx2a+c | eae+/exhA+ | AMP | PYEXHX01.0008 |
| 303191 | 2018 | BD | 2 | C | O157:H7 | Stx2 | stx2a | eae+/exhA+ | – | PYEXHX01.0009 |
As expected, all the strains harbored the wzx/wzy-O157 gene and the fliCH7 gene.
With regard to the virulence profile, all strains harbored stx2, eae and exhA The virulence genotype analysis by WGS allowed the identification of a vast number of virulence factors that are crucial for the pathogenicity mechanism of the strains such as tir, the group of esp proteins (A, B, F, J), katP, nle (A, B), tccP, toxB, among others. Of all the virulence genes present in the database, Figure 1 shows only the results for the genes found for one strain.

In-silico PCR was performed from the FASTA files to determine the Shiga-toxin subtype. Of all nine samples, seven subtyping results agreed with those found by PCR. Two samples identified as stx2a+c by PCR (1903 and 1985) were of the stx2a subtype by in-silico PCR. These two sequences were then analyzed in a complementary manner through the program E. coli Shiga-toxin-typing in the Galaxy platform to verify and confirm results. The 1985 sample was found to be stx2a+c subtype by this method (in agreement with PCR results), while the results for the 1903 sample was again the stx2a subtype.
As for the putative virulence determinants, all the isolates carried ECSP_3620 and ECSP_0242, 8/9 ECSP_2687, 5/9 ECSP_3286, 4/9 ECSP_2870/2872 and 3/9 ECSP_1773 (Fig. 2).

hqSNPs phylogenetic tree and main characteristics of E. coli O157:H7 isolated from human samples in Paraguay.
a The amplicon for the marker gene Z5935 was not predictable by in-silico PCR and hence no lineage was assigned to these isolates.
b The isolates belonged to one of clades 4, 5, 6, 7 or 9 since the subtyping scheme described by Riordan et al. cannot differentiate strains from clade 4, 5, 6, 7, and 9.
AMR analysis identified 7 genes associated with resistance to β-lactamases (blaEC, blaTEM), aminoglycosides (aph(6)-Id, aph(3′)-Id), sulfonamides (sul2) and trimethoprim (dfrA8) among the strains (Fig. 1). Three plasmid replicons sequences were found among the O157:H7 strains corresponding to lncFIB(AP001918), lncFII and lncl1-l (Gamma) incompatibility groups (Fig. 1)
All the strains were found to be ST 11 (allelic profile adk:12 fumC:12 gyrB:8 icd:12 mdh:15 purA:2 recA:2).
As for the LSPA-6 assay, we could not assign lineage to three isolates since the amplicon for the marker gene Z5935 was not predictable by in-silico PCR. Five of the strains belonged to lineage I/II, and one to lineage II. Clade determination was performed according to the algorithm developed by Riordan et al.36. Six out of nine strains (6/9) belonged to the hypervirulent clade 8, and the three others belonged to clades 4–7 or 9 (Fig. 2). Clades 4–7 and 9 strains share the same SNP profile for the target genes in the clade typing method described by Riordan et al.36; therefore, they cannot be differentiated by this method.
The phylogenetic tree obtained by mapping our nine E. coli O157:H7 paired-end reads against a reference genome showed the genetic diversity of the strains, with strains 303191 and 13316 being the most phylogenetically distant (638 SNPs). However, strains 1903 and 1985 (collected in the same year) appeared to be closely related (1 SNP), thus confirming the results found by PFGE. Moreover, we could observe that strains belonging to clade 8 clustered together, supporting the results of the in-silico PCR for clade detection (Fig. 2).
We further investigated the genomic relatedness between our E. coli O157:H7 strains and other E. coli O157:H7 isolates from humans worldwide. Results showed that the Paraguayan strains are genotypically diverse, and are not especially closely related to strains from other specific locations (Fig. 3).
Discussion
The objective of this study was to achieve a genomic characterization of isolated strains of E. coli O157:H7 in Paraguay by performing the WGS analysis. We decided to investigate STEC O157:H7 STEC strains from clinical cases because they are one of the most prevalent and virulent serotypes associated with severe human diseases such as HUS17–19,42. A study conducted in 2017 found that the predominant causative agent in bacterial diarrhea in Paraguay was diarrheagenic E. coli, which is responsible for 13.4% of cases12,44. Epidemiological data for the first quarter of 2021 showed that diarrheagenic E. coli continues to top the list of bacterial agents that cause diarrheal disease with a 14% prevalence (unpublished data-CPHL).
There are no extensive publications on HUS in Paraguay. The first documented case dates from 1987, with no description of the etiological agents. It was not until 2009 when a clinical case of HUS due to E. coli O157:H7 Stx2 was documented for the first time32. Since 2002, when the STEC surveillance started, it was noted that STEC in diarrhea appears to be relatively low in Paraguay (0.6–0.8%). However, the monitoring and characterization of STEC are essential due to its high pathogenic power. In fact, all our O157:H7 strains were confirmed to have a high pathogenic potential, as they all harbor the stx2, eae, and exhA genes. It has been shown that the strains encoding for stx2 are more virulent and are associated with more severe disease than those encoding stx110, and also that intimin (eae), as well as enterohaemolysin (exhA) play a significant role in STEC pathogenicity29. Moreover, WGS allowed to identify many other virulence genes implicated in adhesion (toxB, iha), survival (chuA, katP, gad, iss, ompT), transduction (esp), secretion (etpD), as well as important effectors (nle, chuA, espP, tir), and toxins (astA) from one single analysis. Besides stx2, eae and exhA; all strains harbored nle genes. Another important virulence factor found in all strains was espP, which might be implicated in the pathogenesis of bloody diarrhea, as EspPα can cleave human coagulation factor V. Further investigation regarding the subtype of the espP gene is necessary since EspPβ and EspPδ are impaired in their autotransporter or proteolytic activity5,24,33. Regarding the Stx subtype, stx2c and stx2a+c were the most prevalent. Subtype stx2a was found exclusively among clade 8 strains. These subtypes are, in fact, more frequently associated with human disease28. It is important to point out that WGS analysis failed to identify stx2c from one strain that harbored stx2a. This situation was previously reported by others2,11, and it may be due to the limitations in de novo assembly from short reads because the regions of variation between these subtypes (stx2a and stx2c) are concentrated at the 5′ and 3′ ends of the coding DNA sequence (CDS), with a largely homogenous region in the center2. With regard to AMR, 4 isolates carried the blaTEM gene and 3 were resistant to ampicillin. One of the strains carrying the blaTEM gene also carried genes that confer resistance to aminoglycosides, sulfonamides, and trimethoprim. Antibiotic susceptibility tests of this strain revealed resistance to ampicillin and trimethoprim–sulfamethoxazole. Interestingly, this strain is the only one with the presumption of carrying the IncFII plasmid incompatibility group.
Although the antibiotic treatment of STEC infections in humans is not recommended, AMR represents a severe threat, especially in highly pathogenic strains, since its ability to remain in the environment and/or animals is increased, thus facilitating its spread.
Regarding the epidemiological characteristics, all the strains studied were from ST 11. This ST was widely isolated worldwide from humans, food, and the environment (EnteroBase). The LSPA-6 assay revealed that none of the strains was from lineage I, which was initially thought to be more frequently associated with human isolates24,45,47. Concerning clade designation, a dominance of hypervirulent clade 8 (6/9 strains) was observed, coinciding with other researchers’ findings in Argentina34,41. Furthermore, the two HUS cases were caused by strains belonging to clade 8. As for the putative virulence factors, ECS_3620, ECS_0242 and ECS_2687 were present in strains from clade 8 and non-8, while ECS_3286, ECS_1773 and ECS_2870/2872 were present in strains from clade 8 exclusively.
The phylogenetic tree obtained with the subset of E. coli O157:H7 strains from Paraguay and 50 closest isolates worldwide showed that while the Paraguayan O157:H7 strains are rather homogeneous regarding clade and lineage assignation, they are genetically diverse. We observed that they are mainly spread among different clusters contrary to Australia's, Argentine's, New Zealand's chosen subsets of samples that appeared to be more homogenous.
ConclusionsWGS has become an increasingly attractive tool for foodborne disease surveillance worldwide since its numerous advantages, such as enhanced resolution and time saving features, enable a broad characterization using a single process. Therefore, implementing WGS in Paraguay will allow us to identify, characterize and subtype foodborne pathogens and support an appropriate outbreak response by tracing the source of infection and elucidating transmission pathways, which is very important to identify potential intervention points. Through WGS we achieved an exhaustive molecular characterization of E. coli O157:H7 strains circulating in Paraguay for almost 20 years. For the first time in Paraguay, we describe the circulation of strains belonging to lineage I/II and clade 8. All strains harbored the eae, exhA and stx2 genes. These characteristics describe the O157:H7 strains isolated from humans in Paraguay as a high risk for public health.
FundingThis work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestThe authors declare that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was carried out with the close collaboration of the genomics department and physiopathogenesis service of the Malbrán Institute – Buenos Aires – Argentina.
The following are the supplementary data to this article:
