





Infectious bovine keratoconjunctivitis (IBK) is an ocular disease that affects bovines and has significant economic and health effects worldwide. Gram negative bacteria Moraxella bovis and Moraxella bovoculi are its main etiological agents. Antimicrobial therapy against IBK is often difficult in beef and dairy herds and, although vaccines are commercially available, their efficacy is variable and dependent on local strains. The aim of this study was to analyze for the first time the genomes of Uruguayan clinical isolates of M. bovis and M. bovoculi. The genomes were de novo assembled and annotated; the genetic basis of fimbrial synthesis was analyzed and virulence factors were identified. A 94% coverage in the reference genomes of both species, and more than 80% similarity to the reference genomes were observed. The mechanism of fimbrial phase variation in M. bovis was detected, and the tfpQ orientation of these genes confirmed, in an inversion region of approximately 2.18kb. No phase variation was determined in the fimbrial gene of M. bovoculi. When virulence factors were compared between strains, it was observed that fimbrial genes have 36.2% sequence similarity. In contrast, the TonB-dependent lactoferrin/transferrin receptor exhibited the highest percentage of amino acid similarity (97.7%) between strains, followed by cytotoxins MbxA/MbvA and the ferric uptake regulator. The role of these virulence factors in the pathogenesis of IBK and their potential as vaccine components should be explored.
La queratoconjuntivitis infecciosa bovina (QIB) es una enfermedad ocular que afecta al ganado, con impacto significativo en todo el mundo. Las bacterias Moraxella bovis y Moraxella bovoculi son sus principales agentes etiológicos. La terapia antimicrobiana contra la QIB suele ser dificultosa en el campo y, aunque existen vacunas, su eficacia es variable y dependiente de las cepas que circulan localmente. El objetivo de este estudio fue analizar por primera vez los genomas de aislamientos clínicos de M. bovis y M. bovoculi en Uruguay. Los genomas se ensamblaron de novo y se anotaron; además, se analizaron las bases genéticas de la síntesis de fimbrias y se identificaron factores de virulencia. Se alcanzó una cobertura del 94% de los genomas de referencia de ambas especies y la similitud con ellos fue superior al 80%. Se encontró el mecanismo de variación de fase fimbrial en M. bovis, y se confirmó la orientación tfpQ de dichos genes en una región de inversión de aproximadamente 2,18kb. No se determinó variación de fase en el gen fimbrial de M. bovoculi. Al comparar los factores de virulencia entre cepas, se observó que los genes fimbriales presentan solo un 36,2% de similitud de secuencia. Por el contrario, el receptor de lactoferrina/transferrina dependiente de TonB presentó el mayor porcentaje de similitud a nivel de aminoácidos (97,7%), seguido de las citotoxinas MbxA/MbvA y del regulador de captación férrica. Debe explorarse el papel de estos factores de virulencia en la patogénesis de la QIB y su potencial como componente de vacunas.
Infectious bovine keratoconjunctivitis (IBK) is a common ocular disease of bovines, generally associated with the Gram negative bacterium Moraxella bovis14. In 2007, a new species, Moraxella bovoculi was isolated from animals with IBK in the USA6, and was later detected in other countries, including Uruguay29. Nowadays, although some authors discuss the causative role of M. bovoculi in the disease, it is being isolated from IBK cases even more frequently than M. bovis16.
Several virulence factors have been associated with the pathogenicity of M. bovis, including fimbriae (also known as pili), lipopolysaccharides (LPS), phospholipase B (whose enzymatic activity generates cell lysis), ferric uptake systems, outer membrane protein (Omp) TolC (involved in the export of small molecules across the outer membrane4,23,24), Omp79 (involved in iron uptake)33 and Omp-CD (adhesin)1. Additionally, M. bovis produces an RTX cytotoxin (MbxA), responsible for cell lysis, constituting one of its most studied virulence factors. This cytotoxin is expressed in hemolytic M. bovis strains4,5,23. On the other hand, a similar variant of the M. bovis cytotoxin is also expressed by M. bovoculi (MbvA)2,5. Its contribution to the pathogenesis of IBK has been proved, whereas the significance of M. bovoculi pilin in the pathogenesis of IBK is currently in debate. Recent studies have suggested that M. bovoculi pilin would be more conserved than M. bovis pilin, and a role in colonizing the bovine eye is presumed3.
Type IV fimbriae are the most studied M. bovis virulence factors; they mediate adhesion to the bovine corneal surface, improving the ability to overcome the host defense mechanisms25. It has been shown that M. bovis can express two functionally different types of fimbriae, called type four pili I (TfpI) of 20kDa, and type four pili Q (TfpQ) of 17kDa, as a result of a phase shift mechanism, supported by the inversion of a 2.1kb DNA segment by site-specific recombination. There is an oscillating mechanism of chromosomal rearrangement that limits the expression of one type of fimbriae to the benefit of the other type, since only one promoter is present. Within this 2.1kb region of DNA there are two genes, initially called ORF (open reading frame)-1 and ORF-2. Later, they were named tfpB (ORF-1), whose function remains unknown and is located within the recombinant region, and piv (ORF-2) which encodes a recombinase or invertase responsible for the inversion of this DNA region11. The capacity of M. bovis to produce more than one type of fimbriae increases its ability to evade the host defenses since switching from one type to another could confer antigenic heterogeneity, impairing the effectiveness of the immune response23. On the other hand, M. bovoculi sequenced genomes have shown the lack of this inversion site. Consequently, it would only express one type of fimbriae, which could be more conserved than its M. bovis counterpart3.
Many vaccines against IBK contain inactivated whole bacteria of different M. bovis and M. bovoculi strains or purified fimbriae of M. bovis. These vaccines have shown partial protection; therefore, they are not entirely effective9,20. It is assumed that this limited protection may be due to intraspecific diversity among M. bovis strains17,21. This irregular protection conferred by vaccines against IBK has motivated several authors to investigate the biology of the etiologic agents associated with the disease7. In addition to purified fimbriae, other antigens such as the RTX cytotoxin12 or Omp-CD1 have been proposed as vaccine components. Although specific antibody responses have been observed in preliminary trials, further studies are required to improve this strategy and confirm whether the inclusion of these antigens can prevent the disease.
Based on these considerations and taking into account that the effectiveness of vaccines requires the knowledge of local circulating strains, the genomic analysis of strains isolated from clinical local cases becomes relevant for the design of new IBK prevention strategies. In the present study, the genetic bases of the synthesis of fimbriae and other potential virulence factors of M. bovis and M. bovoculi linked to IBK outbreaks were analyzed using genomic research tools.
Materials and methodsBacterial strains and growth conditionsTwo Moraxella spp. strains collected from eye discharges of calves with IBK symptoms were used. M. bovis EV345 and M. bovoculi PRO strains were collected in 2006 and 2007 respectively, identified and characterized by our group29. Both strains exhibited β-hemolytic activity when grown on Brain Heart Infusion Agar supplemented with 5% ovine blood29,30. They were routinely cultured on this medium at 37°C for 24h or in Brain Heart Infusion (BHI) (at the same conditions) when broth cultures were needed.
DNA extraction and sequencingGenomic DNA of both strains was extracted from fresh bacterial cultures in BHI broth, using the commercial Genomic-Tip 100G kit (Qiagen). Prior to use, DNA purity and concentration were evaluated using Nanodrop (Thermo), and DNA integrity running aliquots on an agarose gel stained with GelRed (Biotum).
The total M. bovis EV345 and M. bovoculi PRO genomic DNAs were sequenced by the service provided by MicrobesNG (https://microbesng.uk/, UK), using Illumina technology.
Data availabilityRaw reads of M. bovis EV345 and M. bovoculi PRO genomes have been deposited at NCBI/SRA under the project accession number PRJNA836137.
Data processing and bioinformatic analysesFiltered raw reads were assessed for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The analysis procedure for both genomes was carried out in the same way. De novo assemblies were executed using SPAdes8, and their quality were evaluated with QUAST13 and Mummer (https://github.com/mummer4/mummer) using the published Moraxella spp. genomes as a reference17,21. MUMmerplot with ggplot2 was also used to make dot plots against the reference genome. Annotations were achieved using Prokka27, with the reference as a guide. To assess the quality of the recent annotation, BUSCO19 and NCBI-Blast (https://blast.ncbi.nlm.nih.gov/) against the reference proteins were used. Variant calling was performed using BCFtools (https://github.com/samtools/bcftools) and later functionally annotated with the variant effect predictor toolbox, SnpEff22. The aligner Bowtie215 was applied to map the filtered reads with their respective reference genomes for further specific analysis. Artemis10,26 and IGV32 tools were performed for visualization data. Sekulovic et al.’s method28 was applied to detect the fimbriae type in M. bovis. MEGAX software was used to construct a phylogenetic tree. The comparison was inferred using the Maximum Likelihood method and the JTT matrix-based model. Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model and then selecting the topology with a superior log-likelihood value.
Orientation of M. bovis fimbrial genesTo confirm the site-specific inversion in the M. bovis fimbrial locus, primers were designed using the Epp63 available genome. The forward primer (5′-ACGCTCAAAAAGGTTTCACC-3′) is located upstream the recombination site, in a conserved region of the tfpQ or tfpI gene (depending on the orientation) and is the same for both configurations, while the reverse for Q conformation (5′-GGGCGATTCTCTCTACTTC-3′) and I conformation (5′-GAAGTAGAGAATCGCCC-3′) are located in a conserved region of the tfpB gene. A total of 16 M. bovis strains, isolated from IBK cases29, and the reference strain Epp63 M. bovis were evaluated. PCR products were sequenced at Macrogen, and the sequences analyzed with BioEdit, version 7.2.5 and MEGA, version 11.0.10.
ResultsAssembly and annotation of M. bovis and M. bovoculiA total of 913094 and 1252392 filtered reads were used to perform de novo assemblies of M. bovis EV345 and M. bovoculi PRO genomes. The assembly process led to 167 contigs for M. bovis and 58 contigs for M. bovoculi (Suppl. Table 1) with 94% coverage with respect to their reference genomes size, suited N50 values and expected GC content. Furthermore, 2815 protein-coding genes and 45 tRNAs for M. bovis, and 2002 protein-coding genes and 43 tRNAs for M. bovoculi were identified (Suppl. Table 1).
Dot plots were performed to compare the assemblies to the reference genomes, showing that they are mainly complete (Suppl. Fig. 1). Nonetheless, some imperfections became apparent in the assemblies. Specifically, small gaps were discerned in the M. bovis assembly, likely stemming from limitations in the sequencing process, where short reads could not comprehensively cover all bases, resulting in the insertion of ‘N's by the assembler (indicated by red arrows in Suppl. Fig. 1). Additionally, a fragmented contig was identified, spanning two distant regions within the reference chromosome (highlighted by red circles in Suppl. Fig 1A). This type of error is a common occurrence in the de novo assembly process. In contrast, the assembly continuity for the M. bovoculi genome appeared to be more favorable (Suppl. Fig. 1B). However, gaps persisted, and small, fragmented contigs were also detected.
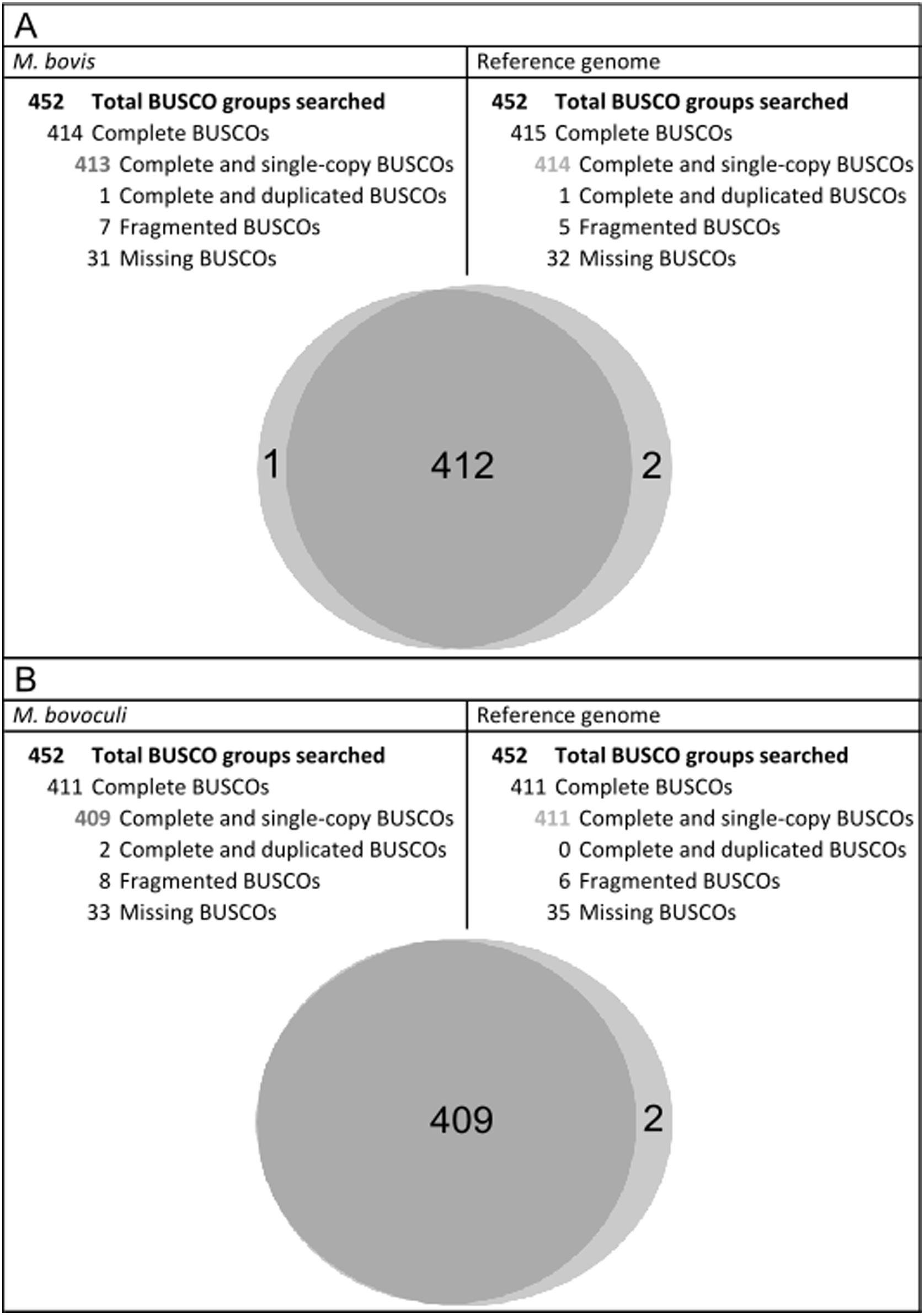
Additionally, BUSCO was run to search universal single-copy orthologous genes in reference strains and in M. bovis EV345 and M. bovoculi PRO genomes. Almost the same complete and single-copy BUSCO groups were detected in both, reference and annotated M. bovis and M. bovoculi strains. Compared to the reference, the process for the assembled M. bovoculi genome resulted in more fragmented genes but fewer missing core gene groups (Fig. 1).
. The same interpretation applies for M. bovoculi (B).")
Benchmarking universal single-copy orthologs of assembled M. bovis and M. bovoculi genomes and their reference for Gamma-proteobacteria class. Results of BUSCO run for M. bovis and M. bovoculi annotated genomes compared to their reference. Top: summary of BUSCO groups found for M. bovis. Bottom: Venn diagram showing the complete and single-copy BUSCOs shared by M. bovis and its reference genome (A). The same interpretation applies for M. bovoculi (B).
Variable positions were searched using the previously published M. bovis and M. bovoculi reference genomes and 10321 and 26938 variants were found respectively, running SnpEff. Among them, 10040 for M. bovis and 26471 for M. bovoculi were identified as single-nucleotide polymorphisms (SNPs). Structural variants were also found. A total of 147 small insertions and 134 deletions were identified in the M. bovis genome, while M. bovoculi showed 267 insertions and 200 deletions (Suppl. Tables 2 and 3).
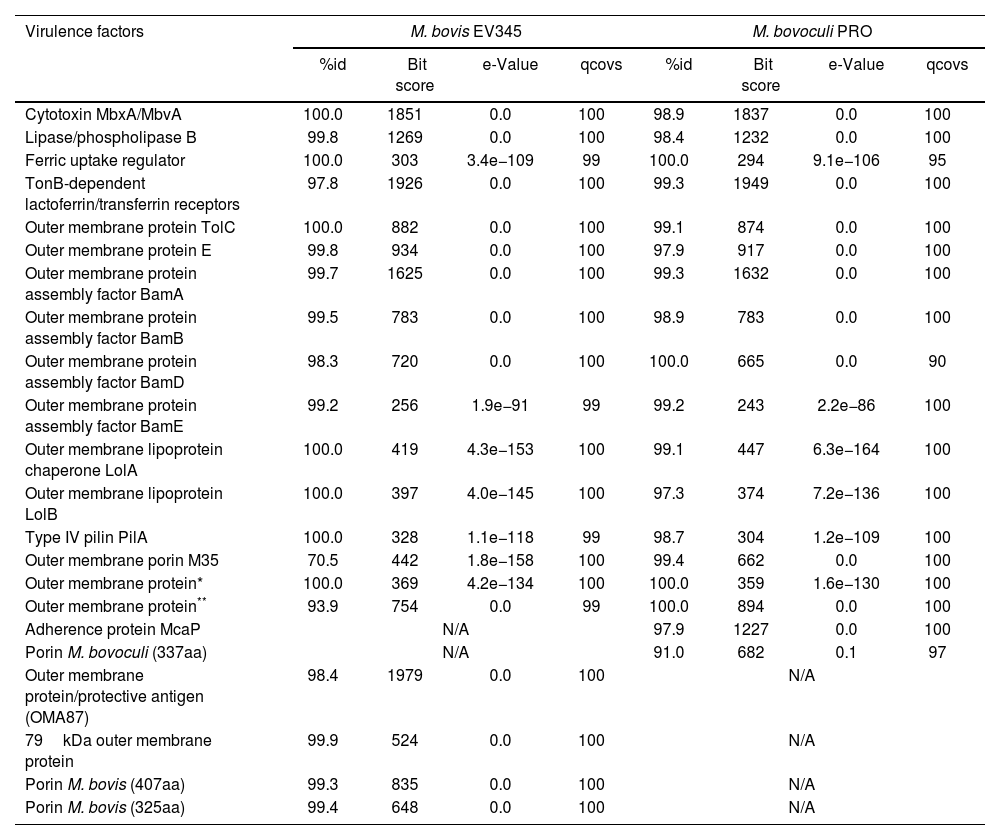
M. bovis EV345 and M. bovoculi PRO virulence profileWith regard to virulence, BLASTp identified a set of potential virulence factors for both species with more than 91% similarity to M. bovis Epp63 and M. bovoculi BAA1259 reference genomes respectively (Table 1), except for M. bovis Omp-M35 which reached 70.5% similarity with respect to the reference strain. Among the identified virulence factors, different Omps, iron uptake proteins, adhesins and toxins, including MbxA and MbvA cytotoxins (Table 1) stood out. Interestingly, cytotoxin MbxA, TolC, lipoproteins LolA and LolB, a ferric uptake regulator and PilA of M. bovis EV345 were identical to the reference Epp63 at the aminoacid level. On the other hand, M. bovoculi PRO outer membrane protein BamD and a ferric uptake regulator and two non-anottated Omps were the only proteins that reached 100% coverage, being identical at the aminoacid level to the reference strain BAA1259 (Table 1).
Presence of virulence factors in the assembled genomes and their similarity to their reference genomes.
| Virulence factors | M. bovis EV345 | M. bovoculi PRO | ||||||
|---|---|---|---|---|---|---|---|---|
| %id | Bit score | e-Value | qcovs | %id | Bit score | e-Value | qcovs | |
| Cytotoxin MbxA/MbvA | 100.0 | 1851 | 0.0 | 100 | 98.9 | 1837 | 0.0 | 100 |
| Lipase/phospholipase B | 99.8 | 1269 | 0.0 | 100 | 98.4 | 1232 | 0.0 | 100 |
| Ferric uptake regulator | 100.0 | 303 | 3.4e−109 | 99 | 100.0 | 294 | 9.1e−106 | 95 |
| TonB-dependent lactoferrin/transferrin receptors | 97.8 | 1926 | 0.0 | 100 | 99.3 | 1949 | 0.0 | 100 |
| Outer membrane protein TolC | 100.0 | 882 | 0.0 | 100 | 99.1 | 874 | 0.0 | 100 |
| Outer membrane protein E | 99.8 | 934 | 0.0 | 100 | 97.9 | 917 | 0.0 | 100 |
| Outer membrane protein assembly factor BamA | 99.7 | 1625 | 0.0 | 100 | 99.3 | 1632 | 0.0 | 100 |
| Outer membrane protein assembly factor BamB | 99.5 | 783 | 0.0 | 100 | 98.9 | 783 | 0.0 | 100 |
| Outer membrane protein assembly factor BamD | 98.3 | 720 | 0.0 | 100 | 100.0 | 665 | 0.0 | 90 |
| Outer membrane protein assembly factor BamE | 99.2 | 256 | 1.9e−91 | 99 | 99.2 | 243 | 2.2e−86 | 100 |
| Outer membrane lipoprotein chaperone LolA | 100.0 | 419 | 4.3e−153 | 100 | 99.1 | 447 | 6.3e−164 | 100 |
| Outer membrane lipoprotein LolB | 100.0 | 397 | 4.0e−145 | 100 | 97.3 | 374 | 7.2e−136 | 100 |
| Type IV pilin PilA | 100.0 | 328 | 1.1e−118 | 99 | 98.7 | 304 | 1.2e−109 | 100 |
| Outer membrane porin M35 | 70.5 | 442 | 1.8e−158 | 100 | 99.4 | 662 | 0.0 | 100 |
| Outer membrane protein* | 100.0 | 369 | 4.2e−134 | 100 | 100.0 | 359 | 1.6e−130 | 100 |
| Outer membrane protein** | 93.9 | 754 | 0.0 | 99 | 100.0 | 894 | 0.0 | 100 |
| Adherence protein McaP | N/A | 97.9 | 1227 | 0.0 | 100 | |||
| Porin M. bovoculi (337aa) | N/A | 91.0 | 682 | 0.1 | 97 | |||
| Outer membrane protein/protective antigen (OMA87) | 98.4 | 1979 | 0.0 | 100 | N/A | |||
| 79kDa outer membrane protein | 99.9 | 524 | 0.0 | 100 | N/A | |||
| Porin M. bovis (407aa) | 99.3 | 835 | 0.0 | 100 | N/A | |||
| Porin M. bovis (325aa) | 99.4 | 648 | 0.0 | 100 | N/A | |||
Known virulence factors found in M. bovis (left) and M. bovoculi (right) assembled genomes. The sequences of the proteins at the aminoacid level were extracted from GenBank and compared to the protein repertoire of the annotated genomes by BLAST alignment with no similarity limitations. Similarity percentage, e-value, bit score and coverage percentage are detailed.
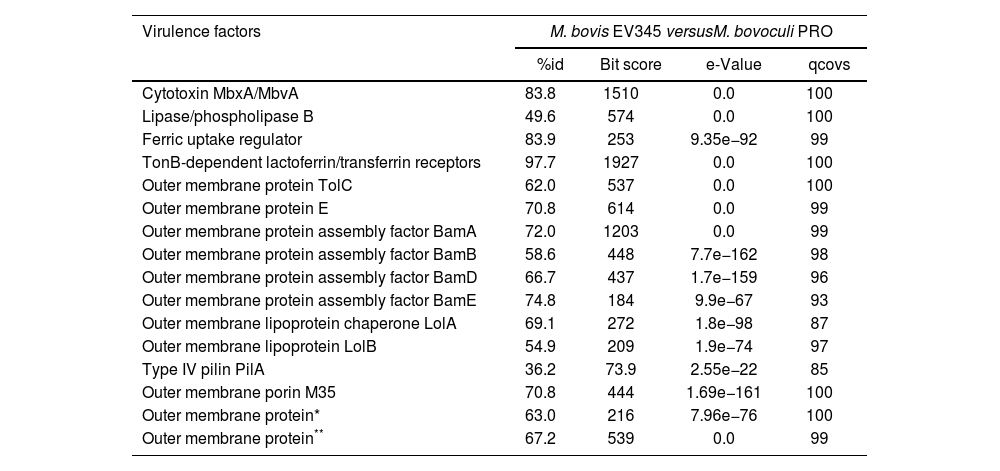
In addition to the comparison with reference strains, selected virulence factors of M. bovis EV345 and M. bovoculi PRO were compared between each other (Table 2). BLASTp results showed that the virulence factors were dissimilar between species. Among the 16 proteins, the TonB-dependent lactoferrin/transferrin receptor was the most conserved Omp between M. bovis and M. bovoculi assembled genomes (97.7%), followed by the ferric uptake regulator and the cytotoxin MbxA/MbvA, both exceeding 83% similarity between strains (Table 2). The remaining virulence factors showed lower similarities, which ranged from 36.2% to 74.8%. The fimbrial PilA protein was the most dissimilar among the sequenced strains (36.2%), followed by the lipase/phospholipase B protein, LolB and BamB with similarities of 49.6%, 54.9% and 58.6%, respectively, between species (Table 2).
Comparison of virulence factors between M. bovis EV345 and M. bovoculi PRO assembled genomes.
| Virulence factors | M. bovis EV345 versusM. bovoculi PRO | |||
|---|---|---|---|---|
| %id | Bit score | e-Value | qcovs | |
| Cytotoxin MbxA/MbvA | 83.8 | 1510 | 0.0 | 100 |
| Lipase/phospholipase B | 49.6 | 574 | 0.0 | 100 |
| Ferric uptake regulator | 83.9 | 253 | 9.35e−92 | 99 |
| TonB-dependent lactoferrin/transferrin receptors | 97.7 | 1927 | 0.0 | 100 |
| Outer membrane protein TolC | 62.0 | 537 | 0.0 | 100 |
| Outer membrane protein E | 70.8 | 614 | 0.0 | 99 |
| Outer membrane protein assembly factor BamA | 72.0 | 1203 | 0.0 | 99 |
| Outer membrane protein assembly factor BamB | 58.6 | 448 | 7.7e−162 | 98 |
| Outer membrane protein assembly factor BamD | 66.7 | 437 | 1.7e−159 | 96 |
| Outer membrane protein assembly factor BamE | 74.8 | 184 | 9.9e−67 | 93 |
| Outer membrane lipoprotein chaperone LolA | 69.1 | 272 | 1.8e−98 | 87 |
| Outer membrane lipoprotein LolB | 54.9 | 209 | 1.9e−74 | 97 |
| Type IV pilin PilA | 36.2 | 73.9 | 2.55e−22 | 85 |
| Outer membrane porin M35 | 70.8 | 444 | 1.69e−161 | 100 |
| Outer membrane protein* | 63.0 | 216 | 7.96e−76 | 100 |
| Outer membrane protein** | 67.2 | 539 | 0.0 | 99 |
Known virulence factors found in M. bovis (left) and M. bovoculi (right) assembled genomes. The sequences of the proteins at the aminoacid level were first extracted from GenBank, searched in the assembled genomes and then the sequences between strains were aligned by BLAST without similarity limitations. Similarity percentage, bit score, e-value and coverage percentage are detailed.
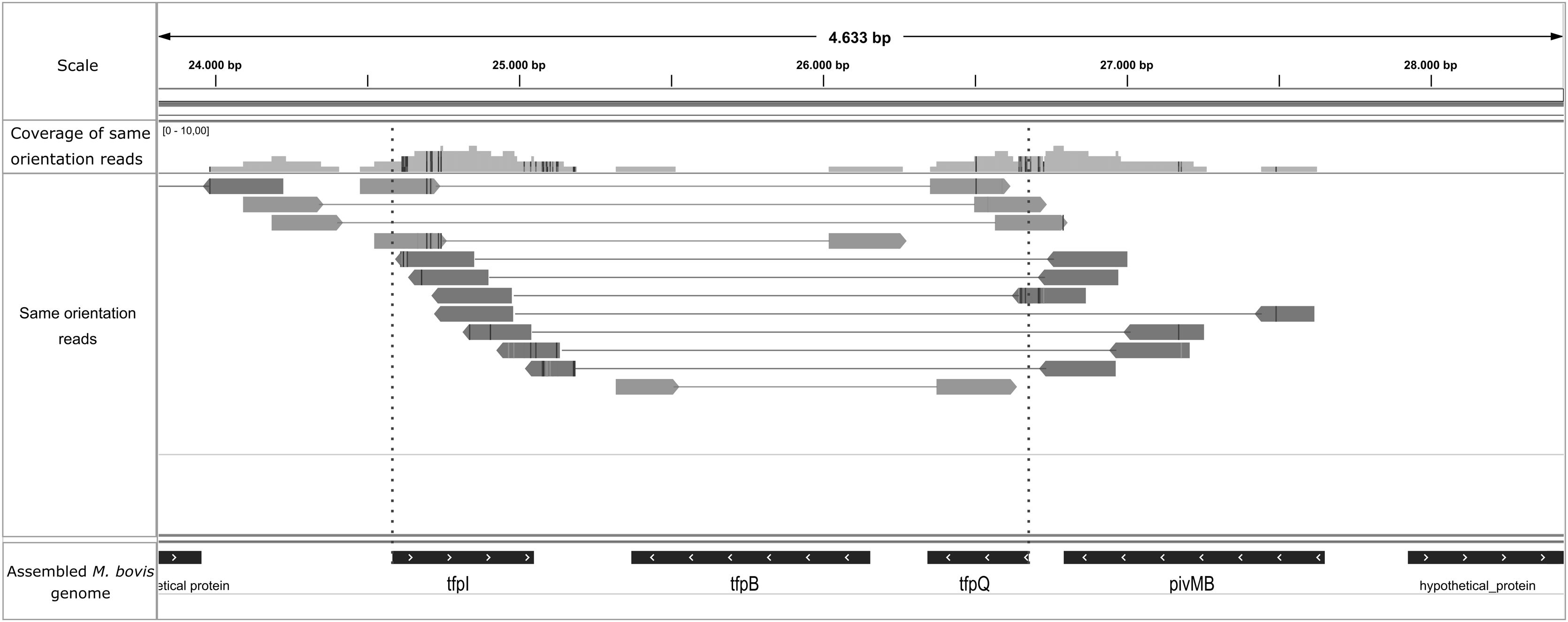
Manual inspection of the M. bovis assembly using Artemis allowed the visualization of fimbrial gene orientation: tfpQ, tfpI, tfpB and pivMB (Suppl. Fig. 2). Based on this arrangement, the tfpQ gene would be in the position of expression of Q fimbriae, whereas the tfpI is inverted (corresponding to a non-expression status of I fimbriae). According to previously reported data, the size of this region where the inversion occurs, was about 2.18kb. The inversion site was suggested with the method provided for Sekulovic et al.28. The high-throughput sequencing reads were aligned to the assembled genome and this atypical group of mapped paired reads was filtered using a simple pipeline. The reads were filtered with the same orientation in this M. bovis genome region (Fig. 2) but not in M. bovoculi, as has been previously reported. Since filtering is possible only in a particular scenario (when one read from a pair map to the invariable region of the genome and the other read pair maps to the invertible segment), the final number of filtered mapping reads is expected to be very low28, which was corroborated in our assays.
.")
Genomic inversion site represented by an unusual clustering of mapped reads in the assembled M. bovis genome. Graphical confirmation of the inversion site in the pilin locus of M. bovis using IGV. Quality reads were mapped to the M. bovis assembled genome and same orientation reads were filtered through the Sekulovic pipeline28. Paired reads are joined by a line and placed just where recombination should occur (dotted red line, above tfpI and tfpQ genes).
In their study, Sekulovic et al.28 also described a simple way to predict the recombination site applying soft clipping while mapping the reads to the genome and extracting the 5-prime end clipped reads28. Using the soft clipping aligner function, the start position of the 5-prime clipped reads mapped, therefore allowing for precise identification of the site where the DNA strand is broken by the enzymatic activity of the recombinase to initiate the strand exchange required for inversion28. Following this pipeline, just a few reads with these features were identified, indicating a possible site-specific recombination site (Fig. 3).
 were extracted through the Sekulovic pipeline28. We identified five reads (highlighted with arrows) that were clipped in the expected region where recombination occurs.")
Putative recombination site represented by 5-prime clipped reads mapped in assembled M. bovis genome. Graphical confirmation of the recombination site in the pilin locus of M. bovis using IGV. Quality reads were mapped to the M. bovis assembled genome and 5-prime end clipped reads (bottom panel) were extracted through the Sekulovic pipeline28. We identified five reads (highlighted with arrows) that were clipped in the expected region where recombination occurs.
We performed a BLASTn analysis between the sequence of our assembled M. bovoculi PilA and 94 other sequences of PilA from the study by Angelos et al.3. The deduced amino acid sequences of these M. bovoculi isolates encoded ten unique PilA sequences that were designated as M. bovoculi PilA groups, A to J. It was found that our assembled PilA had 99% similarity (457 of 459 nt in common) with an isolate from the group C (MT333727.1). This comparison reached the best bit scores in the alignment. When comparing the deduced amino acid sequence of our assembled PilA with those from consensus PilA groups A–J, using a Muscle alignment, we observed a resemblance between our assembled PilA and those from group B and C; sharing with both only a difference of a single amino acid (Fig. 4). To confirm these results, we constructed a phylogenetic tree using the Maximum Likelihood method with the previous alignment (Suppl. Fig. 3). As expected, groups C and B are the nearest to our assembled PilA.
 and consensus M. bovoculi PilA groups A-J isolates derived from cattle with IBK in Western USA3. Differences between groups and the consensus sequence are identified. At residue 61 there was a 50/50 split among the 10 PilA group A-J isolates with deduced amino acid S versus N.")
Alignment of M. bovoculi PilA from the assembled genome and the 10 PilA group isolates. Deduced amino acid sequence aligned by Muscle of M. bovoculi PilA from PRO strain (Assembled PilA) and consensus M. bovoculi PilA groups A-J isolates derived from cattle with IBK in Western USA3. Differences between groups and the consensus sequence are identified. At residue 61 there was a 50/50 split among the 10 PilA group A-J isolates with deduced amino acid S versus N.
The amino acid sequence of the hemolysin MbxA from the assembled M. bovis genome was compared to three hemolysin protein variants recently reported34. Wynn and collaborators found that, among other aspects, amino acid variants determined the associated genotype. Our assembled protein presented one amino acid substitution (K171Q) located closer to the N-terminus of the protein (Suppl. Fig. 4). Since our assembled hemolysin had the Q allele, it would be inferred that M. bovis EV345 corresponds to genotype 2.
Genetic analysis of tfpQ and tfpI fimbriaeBased on the two possible orientations that the M. bovis Epp63 fimbrial genes (GenBank: M32345.1 and GenBank: M59712.1) can adopt and knowing that the TfpB protein gene is conserved and located between them, we designed primers to amplify both conformations. The PCR products contained the coding sequence of the fimbrial gene, the intergenic region, and most of the tfpB gene. The amplification of both genes, the tfpQ and tfpI genes in expression conformation was determined in each strain at the same time (data non-shown). Multiple alignments with MEGA 11.0.10 determined a conserved region toward the 5′, and then all the sequences were highly variable from each other. Further, the non-coding intergenic sequences between the tfpQ or tfpI and the tfpB gene were analyzed. It was determined that the intergenic sequence located between the first fimbrial gene and the tfpB gene is similar between sequences of the same conformation but differs between the Q and I conformations. Based on this fact, this region made it possible to distinguish when the gene was in the tfpQ or tfpI expression conformation (non-shown).
DiscussionM. bovis and M. bovoculi are bovine ocular pathogens responsible for causing IBK6,14. Several factors are involved in IBK pathogenesis, including animal features such as breed and age, environmental factors, management practices, and farm facilities23. However, pathogenicity strongly depends on the intrinsic properties of the specific strains. One strategy to prevent IBK is vaccination. Nowadays, vaccines containing inactivated whole bacteria of different M. bovis and M. bovoculi strains, or purified fimbriae of M. bovis are available9,20. Effectiveness and protection of these vaccines are variable but rarely complete.
In this study, the genomes of two Uruguayan isolates of M. bovis and M. bovoculi obtained from the eyes of calves with clinical IBK signs were sequenced and analyzed for the first time in the country. Comparative genomics using local strains may be important to predict the effectiveness of commercial vaccines.
Complete assemblies reached 94% coverage of both strains reference genomes size. Reciprocal BLASTp and BUSCO analysis of the annotations showed a remarkable resemblance with the reference genomes. It was observed that our annotated proteins contained more than 80% of the genomes for both species based on their repertoire of conserved protein characteristics within their taxonomic class. Genetic variants were searched through VCF files to see how changes in the CDS would affect the gene product. With regard to the variant type, the SnpEff report shows that approximately 97–98% of the variants in both strains are SNPs. While the number of variants may appear high, most of them belong to a silent functional class, where the genes do not have a significant impact. Among the annotated proteins for both assembled genomes, the presence of the most studied virulence factors was observed in both species. All of them showed a high percentage of similarity within their reference genomes (over 90%), except for M. bovis Omp-M35, which had a similarity value of 70.5% compared with reference Epp63. These results allow us to infer that the local M. bovis and M. bovoculi pathogenic strains exhibit virulence attributes similar to those of the reference strains.
In order to assess similarity among the selected virulence factors in the local strains PRO and EV345, the sequences of these assembled proteins were extracted and compared with each other. It could be observed that the TonB-dependent lactoferrin/transferrin receptor carried the top percentage of similarity at the amino acid level among M. bovis and M. bovoculi assembled genomes (97.7%), followed by cytotoxins MbxA/MbvA and the ferric uptake regulator. These observations may be explained since the toxic effect of RTX and the ferric uptake activity are critical for bacterial survival and IBK pathogenesis. An in-depth study of these virulence factors could result in a new vaccine component against IBK. Conversely, it was noted that type IV pilin PilA had 36.2% sequence similarity among both strains. Type IV pilin have been broadly studied and is considered an essential component of vaccine preparations. The similarity value for PilA was comparable to the one obtained by Calcutt et al.21, who determined a 38% pilin sequence homology between both species. The high diversity of this protein suggests that it would be unlikely to have cross responsiveness between species. A specific antibody response for each species would be desirable to counteract IBK; therefore, the inclusion of both antigens within the vaccine components should be considered. With regard to the TolC protein, even though the percentages of similarity between M. bovis EV345 and M. bovoculi PRO were high when the BLASTp sequences were compared with the reference strains (100% and 99.1% respectively), the similarity between species was one of the lowest in our study (62%). Previous studies by our group had already determined this difference between species using a large collection of M. bovis and M. bovoculi IBK-clinical isolates31. Here we observed that TolC is conserved within species but differs when M. bovis and M. bovoculi are compared. The analysis of virulence factors suggests that the inclusion of both M. bovis and M. bovoculi proteins should be considered to provide an effective immune response.
Regarding the fimbrial machinery, the orientation of M. bovis fimbrial genes confirmed the type four pili Q (tfpQ) in the M. bovis EV345 genome, a product of the recombination in this locus. To confirm the presence of genomic inversions in this region and estimate the specific recombination site, we used the method described by Sekulovic et al.28. These authors proposed that site-specific recombination events could be detected by looking for signatures following high-throughput deep sequencing of microbial genomes. Several reads with the same relative orientation and increased inner-mate distance in the fimbria locus region were identified. No other significant groups of atypical-paired reads were found in the rest of the assembled M. bovis genome. Although the number of filtered mapping reads was low, this amount was enough to predict a possible inversion site, as they were significantly more frequent than in other genome places. Moreover, this atypical group of reads found was the only one in the whole genome that satisfied all the characteristics described by Sekulovic et al. to be finally identified as a genomic inversion28. Likewise, the existence of a site-specific inversion zone in the M. bovis fimbrial locus was confirmed by PCR amplification, sequencing, and multiple alignment of the intergenic region between the tfpQ-I and the tfpB genes. Additionally, we were able to identify a putative recombination site, although with the support of a low number of reads. However, considering this filter is even stricter, a much smaller number of reads is expected. We estimated that around 7–8% of the total mapped reads in this region were filtered by 5-prime clipped, suggesting that this might be representing the fimbrial recombination percentage for M. bovis in our data. Nevertheless, this estimated number has an associated error due to the low count of filtered reads observed. Interestingly, 3 out of these 5 reads seem to be cut into the same position, while the other two have a different recombination site a few nucleotides further away. This finding could show that the recombination is not highly accurate, and perhaps the recombination site is lax and variable. As expected, no unusual orientations of mapping paired reads were detected in the M. bovoculi assembled genome. This finding confirms that this species does not have a genomic inversion mechanism and lacks fimbrial phase change mechanisms, confirming previous results21.
Continuing the genomic analysis of M. bovoculi fimbriae, we demonstrated that phylogenetically, and at the amino acid level, there is a great similarity of M. bovoculi PilA from PRO strain with PilA groups B and C, recently identified in IBK-associated M. bovoculi isolates in the USA3. Moreover, BLASTn analyses indicated that the assembled sequence of the pilA gene showed fewer changes compared to an isolate of group C. PilA group C has been reported as one of the most widely distributed among PilA groups3. With the increasing detection frequency of M. bovoculi in reported cases of IBK16, the study of this pathogen and its virulence factors becomes necessary. The role of M. bovoculi fimbriae in developing IBK symptoms is still under investigation. It has been proposed that it would have an important role in the attachment to the ocular surface and survival in the mucosa3. Considering that the biogenesis machinery of M. bovoculi fimbriae lacks the phase change system described for M. bovis and that a limited number of fimbrial groups exist even in distant geographical locations, it could be speculated that M. bovoculi fimbriae would be an interesting target for the improvement of IBK vaccines.
To date, two proposed M. bovis genotypes are based on different genomic and plasmid characteristics34. Among other things, the Clawson group described three variants of the hemolysin MbxA, being variant 2 associated with genotype 2. To identify the genotype of our assembled M. bovis genome, we performed a comparison at the amino acid level between the consensus sequence of the three variants for the hemolysin and our hemolysin from the M. bovis EV345 genome. The amino acid substitutions that were found correspond to genotype 2. It has been proposed that M. bovis genotypes show differences regarding their pathogenesis34. As proposed by Wynn et al.34, genotype 2 strains could have an advantage in the first steps of invasion and adhesion to the corneal surface over those of genotype 1, potentially affecting the pathogenicity and virulence of the strains18,34.
ConclusionsWe were able to correctly de novo assemble and annotate the genomes of IBK-associated M. bovis and M. bovoculi Uruguayan strains. Both genomes, including their virulence genes, were very similar to those already published from distant geographic origins, including genes of Omps and cytotoxins. This could be important for the design of vaccines to be used worldwide. As predicted, a fimbrial phase variation mechanism and type four pili Q (TfpQ) was found in the M. bovis EV345 genome. No other putative recombination sites were found. A putative and presumably lax recombination site was identified in M. bovis using a method based on high-throughput deep sequencing. To our knowledge, this is the first time that such an accurate prediction has been made with high-throughput deep sequencing data for M. bovis. No phase variation mechanisms were found in the M. bovoculi PRO fimbrial gene, whereas the predicted PilA fimbrial protein corresponds to group C.
A series of virulence factors, including Omps, proteins associated with iron uptake, cytotoxins and fimbriae were described as co-ocurring in both M. bovis EV345 and M. bovoculi PRO local strains, although with different levels of sequence homology between species. The TonB-dependent lactoferrin/transferrin receptor was the only virulence factor with over 97% similarity between species. Further studies to evaluate the role of this virulence factor in IBK pathogenesis and its potential as a vaccine component should be explored. The simultaneous inclusion of antigens from both species could also be a way to improve the humoral response and protection of vaccine preparations against IBK, and OMPs, such as TolC, could be a promising alternative considering the similarity among strains of the same species.
FundingThe present work was partially supported by Virbac Uruguay SA.
Conflicts of interestsThe authors declared no potential conflicts of interest.
The following are the supplementary data to this article:
