A previous sequence analysis of a US5 gene fragment of infectious laryngotracheitis virus (ILTV) performed in an Argentinian epidemiological study allowed to differentiate between wild and vaccine strains. This analysis also defined five ILTV haplotypes with specific variations at positions 461, 484, 832, 878 and 894 of the US5 gene. This characterization of viral strains may also be accomplished using the High-Resolution Melting Analysis (HRMA), which has been described as an effective, fast and sensitive method to detect mutations in PCR products. In the present study, an HRM protocol was developed with the aim of characterizing the circulating ILTV strains in Argentina. The specificity of this tool was confirmed in different DNA diluents, without interference from heterologous DNA or other cellular metabolites. Additionally, the salt concentration in the elution buffer used for DNA extraction did not alter the curve profiles. Higher concentrations of DNA (Ct≅26.0) displayed well-defined curve profiles, whereas lower concentrations (Ct≅32.5) exhibited more heterogeneous curves. The HRMA showed 97.49% concordance with the reference technique, i.e., sequencing. The HRM protocol has the capability to perform DNA amplification prior to its characterization. Thus, eventually this technique may be used simultaneously as a diagnostic tool. This advantage implies a significant reduction in the time and effort involved in sample processing.
En un estudio epidemiológico realizado previamente en Argentina, se analizó la secuencia de un fragmento del gen US5 del virus de la laringotraqueítis infecciosa (ILTV), lo que permitió diferenciar las cepas de campo de las vacunales. También esto permitió definir cinco haplotipos del ILTV, con variaciones específicas en las posiciones 461, 484, 832, 878 y 894 del gen US5. La caracterización de las cepas virales también puede lograrse mediante el análisis de la disociación de alta resolución o high-resolution melting analysis (HRMA), descripto como un método efectivo, rápido y sensible para detectar mutaciones en productos de PCR. En el presente estudio se desarrolló un protocolo de disociación de alta resolución con el objetivo de caracterizar cepas del ILTV circulantes en Argentina. Para ello,se confirmó la especificidad de esta herramienta en diferentes diluyentes del ADN de las muestras, sin observarse interferencias en presencia de ADN heterólogo u otros metabolitos celulares. Asimismo, la concentración de sales en el buffer de elución utilizado durante la extracción de ADN no alteró los perfiles de las curvas. Se obtuvieron perfiles bien definidos con concentraciones de ADN más elevadas (Ct ≅ 26.0), mientras que concentraciones más bajas presentaron curvas heterogéneas (Ct ≅ 32.5). El HRMA mostró una concordancia del 97.49% con la técnica de referencia, la secuenciación. El protocolo de disociación de alta resolución amplifica el ADN antes de su caracterización, por lo que esta técnica podría ser eventualmente utilizada para confirmar la presencia del ILTV y, al mismo tiempo, distinguir haplotipos, optimizando su valor como herramienta de diagnóstico. Esta característica implica una reducción significativa en el tiempo dedicado al procesamiento de muestras.
Infectious laryngotracheitis (ILT) is a worldwide occurring respiratory chicken disease, generally observed in areas of highly intensive poultry production14.
The causal agent of this disease is the infectious laryngotracheitis virus (ILTV) or Gallid Herpesvirus-1 (GaHV-1), a member of the genus Iltovirus of the family Herpesviridae9, whose genome consists of a linear double stranded DNA of 153 to 155kbp12,18.
ILTV infection is characteristically localized in the ocular conjunctiva and respiratory tract (larynx and trachea) and affects chickens from 14 days of age11. The clinical signs include different grades of conjunctivitis, nasal discharge, coughing and depression. On occasions, more virulent ILTV strains can lead to a more severe form of the disease, with high respiratory distress as a consequence of tracheal obstruction with fibrino-hemorrhagic exudates, causing asphyxia and death14.
The currently available ILT vaccines (live attenuated and vectored) are capable of controlling the disease. However, it was proved that they were not equally efficient against the ILTV challenge29. Usually, those differences can also be observed under field conditions and they might depend not only on the type of vaccine but also on the route of administration and virulence of the field strain7. Thus, despite the extensive use of attenuated and vectored viral vaccines, outbreaks are still common. This situation has motivated several studies aiming at differentiating vaccine strains from field isolates. Diverse genomic regions as well as molecular methodologies have been applied to study circulating variants in several countries. The implemented techniques have evolved from restriction fragment length polymorphism (RFLP) analysis of the entire viral genome to sequence analysis of some ILTV genes1,3–5,8,16,19,20,23.
Recently, our group has demonstrated that the sequence analysis of a fragment of the US5 gene (glycoprotein J or gJ) allowed to characterize Argentinian field strains into five groups (haplotypes), two of them belonging to vaccine strains8. Through this analysis, we detected specific variations at five critical positions over a 1279-bp region of the US5 gene, and therefore stated that five haplotypes circulate in this country.
Mutation scanning may also be accomplished using the High-Resolution Melting Analysis (HRMA). This technique is effective, fast and highly sensitive, and allows to detect punctual mutations along the sequence of PCR products10,22,25,26. In this methodology, the fragment of interest is amplified in the presence of a fluorescent dye at saturating concentrations; subsequently, the PCR product is denatured by gradual temperature increments and, finally, the characterization is achieved by comparing variations in fluorescence as a function of melting point temperatures10,17. Since the melting temperature (Tm) of a product is dependent on the GC content, length, and sequence of the sample, PCR products can be distinguished by their melting curves27. The amplification and subsequent mutation scanning performed in an HRM protocol occur in the same tube, which makes it less laborious and eliminates contamination concerns21,26.
The purpose of the present study was to design and implement an HRM protocol in order to characterize ILTV strains circulating in Argentina and to compare the results with the reference sequencing technique.
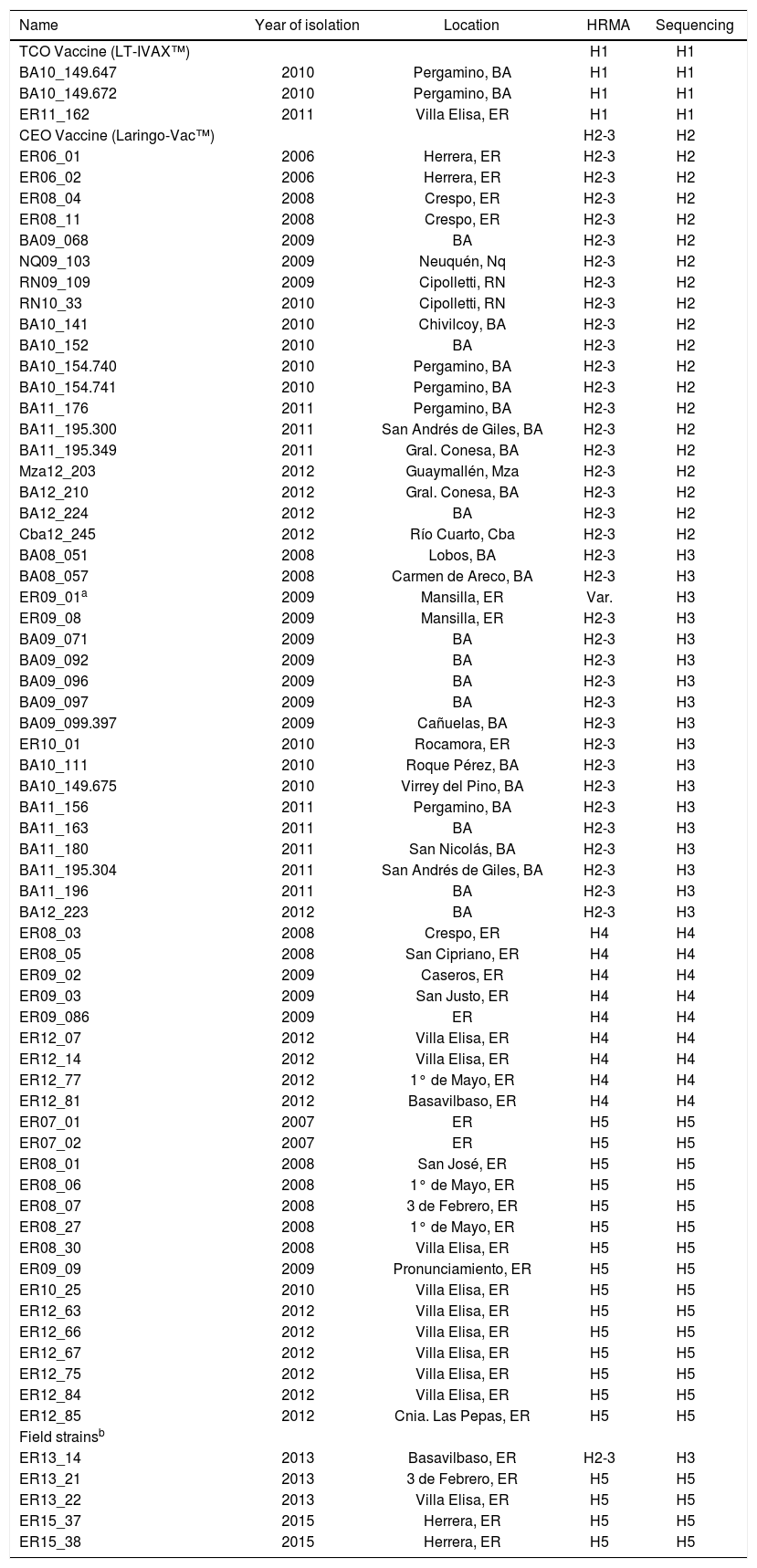
Materials and methodsViral samplesILTV samples were obtained from the tracheas of animals infected during field outbreaks in different locations of Argentina, between 2006 and 2012 (Table 1).
Viral strains characterized by sequencing and HRMA.
| Name | Year of isolation | Location | HRMA | Sequencing |
|---|---|---|---|---|
| TCO Vaccine (LT-IVAX™) | H1 | H1 | ||
| BA10_149.647 | 2010 | Pergamino, BA | H1 | H1 |
| BA10_149.672 | 2010 | Pergamino, BA | H1 | H1 |
| ER11_162 | 2011 | Villa Elisa, ER | H1 | H1 |
| CEO Vaccine (Laringo-Vac™) | H2-3 | H2 | ||
| ER06_01 | 2006 | Herrera, ER | H2-3 | H2 |
| ER06_02 | 2006 | Herrera, ER | H2-3 | H2 |
| ER08_04 | 2008 | Crespo, ER | H2-3 | H2 |
| ER08_11 | 2008 | Crespo, ER | H2-3 | H2 |
| BA09_068 | 2009 | BA | H2-3 | H2 |
| NQ09_103 | 2009 | Neuquén, Nq | H2-3 | H2 |
| RN09_109 | 2009 | Cipolletti, RN | H2-3 | H2 |
| RN10_33 | 2010 | Cipolletti, RN | H2-3 | H2 |
| BA10_141 | 2010 | Chivilcoy, BA | H2-3 | H2 |
| BA10_152 | 2010 | BA | H2-3 | H2 |
| BA10_154.740 | 2010 | Pergamino, BA | H2-3 | H2 |
| BA10_154.741 | 2010 | Pergamino, BA | H2-3 | H2 |
| BA11_176 | 2011 | Pergamino, BA | H2-3 | H2 |
| BA11_195.300 | 2011 | San Andrés de Giles, BA | H2-3 | H2 |
| BA11_195.349 | 2011 | Gral. Conesa, BA | H2-3 | H2 |
| Mza12_203 | 2012 | Guaymallén, Mza | H2-3 | H2 |
| BA12_210 | 2012 | Gral. Conesa, BA | H2-3 | H2 |
| BA12_224 | 2012 | BA | H2-3 | H2 |
| Cba12_245 | 2012 | Río Cuarto, Cba | H2-3 | H2 |
| BA08_051 | 2008 | Lobos, BA | H2-3 | H3 |
| BA08_057 | 2008 | Carmen de Areco, BA | H2-3 | H3 |
| ER09_01a | 2009 | Mansilla, ER | Var. | H3 |
| ER09_08 | 2009 | Mansilla, ER | H2-3 | H3 |
| BA09_071 | 2009 | BA | H2-3 | H3 |
| BA09_092 | 2009 | BA | H2-3 | H3 |
| BA09_096 | 2009 | BA | H2-3 | H3 |
| BA09_097 | 2009 | BA | H2-3 | H3 |
| BA09_099.397 | 2009 | Cañuelas, BA | H2-3 | H3 |
| ER10_01 | 2010 | Rocamora, ER | H2-3 | H3 |
| BA10_111 | 2010 | Roque Pérez, BA | H2-3 | H3 |
| BA10_149.675 | 2010 | Virrey del Pino, BA | H2-3 | H3 |
| BA11_156 | 2011 | Pergamino, BA | H2-3 | H3 |
| BA11_163 | 2011 | BA | H2-3 | H3 |
| BA11_180 | 2011 | San Nicolás, BA | H2-3 | H3 |
| BA11_195.304 | 2011 | San Andrés de Giles, BA | H2-3 | H3 |
| BA11_196 | 2011 | BA | H2-3 | H3 |
| BA12_223 | 2012 | BA | H2-3 | H3 |
| ER08_03 | 2008 | Crespo, ER | H4 | H4 |
| ER08_05 | 2008 | San Cipriano, ER | H4 | H4 |
| ER09_02 | 2009 | Caseros, ER | H4 | H4 |
| ER09_03 | 2009 | San Justo, ER | H4 | H4 |
| ER09_086 | 2009 | ER | H4 | H4 |
| ER12_07 | 2012 | Villa Elisa, ER | H4 | H4 |
| ER12_14 | 2012 | Villa Elisa, ER | H4 | H4 |
| ER12_77 | 2012 | 1° de Mayo, ER | H4 | H4 |
| ER12_81 | 2012 | Basavilbaso, ER | H4 | H4 |
| ER07_01 | 2007 | ER | H5 | H5 |
| ER07_02 | 2007 | ER | H5 | H5 |
| ER08_01 | 2008 | San José, ER | H5 | H5 |
| ER08_06 | 2008 | 1° de Mayo, ER | H5 | H5 |
| ER08_07 | 2008 | 3 de Febrero, ER | H5 | H5 |
| ER08_27 | 2008 | 1° de Mayo, ER | H5 | H5 |
| ER08_30 | 2008 | Villa Elisa, ER | H5 | H5 |
| ER09_09 | 2009 | Pronunciamiento, ER | H5 | H5 |
| ER10_25 | 2010 | Villa Elisa, ER | H5 | H5 |
| ER12_63 | 2012 | Villa Elisa, ER | H5 | H5 |
| ER12_66 | 2012 | Villa Elisa, ER | H5 | H5 |
| ER12_67 | 2012 | Villa Elisa, ER | H5 | H5 |
| ER12_75 | 2012 | Villa Elisa, ER | H5 | H5 |
| ER12_84 | 2012 | Villa Elisa, ER | H5 | H5 |
| ER12_85 | 2012 | Cnia. Las Pepas, ER | H5 | H5 |
| Field strainsb | ||||
| ER13_14 | 2013 | Basavilbaso, ER | H2-3 | H3 |
| ER13_21 | 2013 | 3 de Febrero, ER | H5 | H5 |
| ER13_22 | 2013 | Villa Elisa, ER | H5 | H5 |
| ER15_37 | 2015 | Herrera, ER | H5 | H5 |
| ER15_38 | 2015 | Herrera, ER | H5 | H5 |
Two vaccines were also included in the study, CEO attenuated by several passages in chicken embryo (Laringo-Vac™ Solvay Animal Health) and TCO attenuated by tissue culture passages (LT-IVAX™ Schering Plough).
DNA extractionViral DNA was extracted from tracheal swabs or tracheal homogenates in PBS medium supplemented with antibiotics as described by Craig et al8. Briefly, tracheal mucosa samples were scraped and homogenized with sterile sand and PBS supplemented with penicillin 10000IU/ml, streptomycin 5000μg/ml, gentamicin sulfate 1000μg/ml, kanamycin sulfate 700μg/ml, and amphotericin B 10μg/ml (Sigma Chemical Co™, St. Louis, MO, USA). Subsequently, the homogenates were centrifuged for 1min at 5000g to eliminate cell debris and sand. Tracheal swabs were obtained by a strong rubbing of the mucosal zone and suspended in 2ml of sterile PBS supplemented with antibiotics. Finally, DNA was extracted from the supernatants using the QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, USA), according to the manufacturer's instructions.
PrimersTwo sets of primers (Suppl. Table 3) were designed using “Primer Quest Tool” software (https://www.idtdna.com/site) on sequences previously published in GenBank. These primers were selected avoiding secondary structures (because this may result in unusual melting profiles) and with an annealing temperature of 60°C. These primers amplify two regions within the US5 gene: zone 1 (Z1 of 137bp) and zone 2 (Z2 of 136bp). These two regions together include the five varying nucleotide positions that define the five haplotypes. Three concentrations of the primers were tested (0.1μM, 0.3μM and 0.5μM) to optimize the protocol.
HRMA sensitivity assayA sensitivity assay was performed with a clone containing a 512-bp DNA fragment Z1-Z2 from the TCO vaccine by transforming E. coli DH5α with a “pGEM®-T easy vector” (Promega Co., Fitchburg, WI, USA), following the protocol described by Sambrook28. Differences between results obtained in the presence of heterologous DNA were evaluated by performing serial ten-fold dilutions of the clone (final concentrations of 1010copies/μl to 1copy/μl). The dilutions were prepared in two different diluents (a) nuclease-free water and (b) DNA solution extracted from a negative sample for ILTV (called matrix). Finally, a real time PCR of each dilution was performed in a StepOnePlusTM System (Applied BiosystemsTM, Foster City, CA, USA). The cycling program includes a final denaturation step performed from 60°C to 95°C for 15s, with a temperature rising slope of 0.3%, which corresponds to a variation of about 0.1°C/s in the instrument (Suppl. Table 4). Reaction mixes were prepared in a final volume of 20μl, using “MeltDoctorTM HRM Master Mix” (Applied BiosystemsTM) and 0.3μM of each set of primers (gJZ1-Fw and gJZ1-Rv, and gJZ2-Fw and gJZ2-Rv).
The values of slope and intercept of each obtained standard curve were compared applying Multiple Linear Regression (p<0.05).
HRMA haplotype testFive clones were used as controls for this trial: one for each ILTV haplotype circulating in Argentina. For this purpose, the amplified Z1-Z2 fragment was cloned from the TCO vaccine (haplotype 1), the CEO vaccine (haplotype 2) and field samples BA12_223 (haplotype 3), ER09_02 (haplotype 4) and ER13_122 (haplotype 5). These control clones are henceforth called H1, H2, H3, H4 and H5, depending on the haplotype to which they belong. Then, serial ten-fold dilutions were prepared for each clone separately using (a) nuclease-free water and (b) the provided elution buffer (QIAamp DNA Mini Kit, Qiagen Inc.) to determine if the presence of salts in the buffer could interfere with resultant profiles. Dilutions with 104 and 102 DNA copies/μl in both media were selected (Ct=32.5 and 26.0, respectively, based on the results of the sensitivity assay) and analyzed in triplicate by HRM. PCR conditions were similar to those of the sensitivity assay, except for the last denaturation step, when the temperature increase was achieved on a continuous mode with the addition of a final step (Suppl. Table 4).
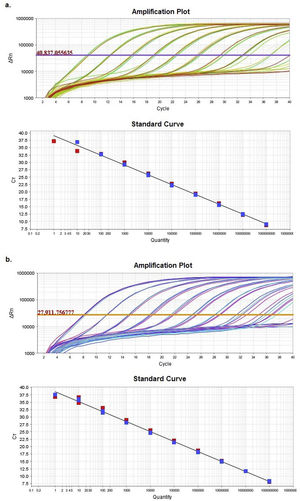
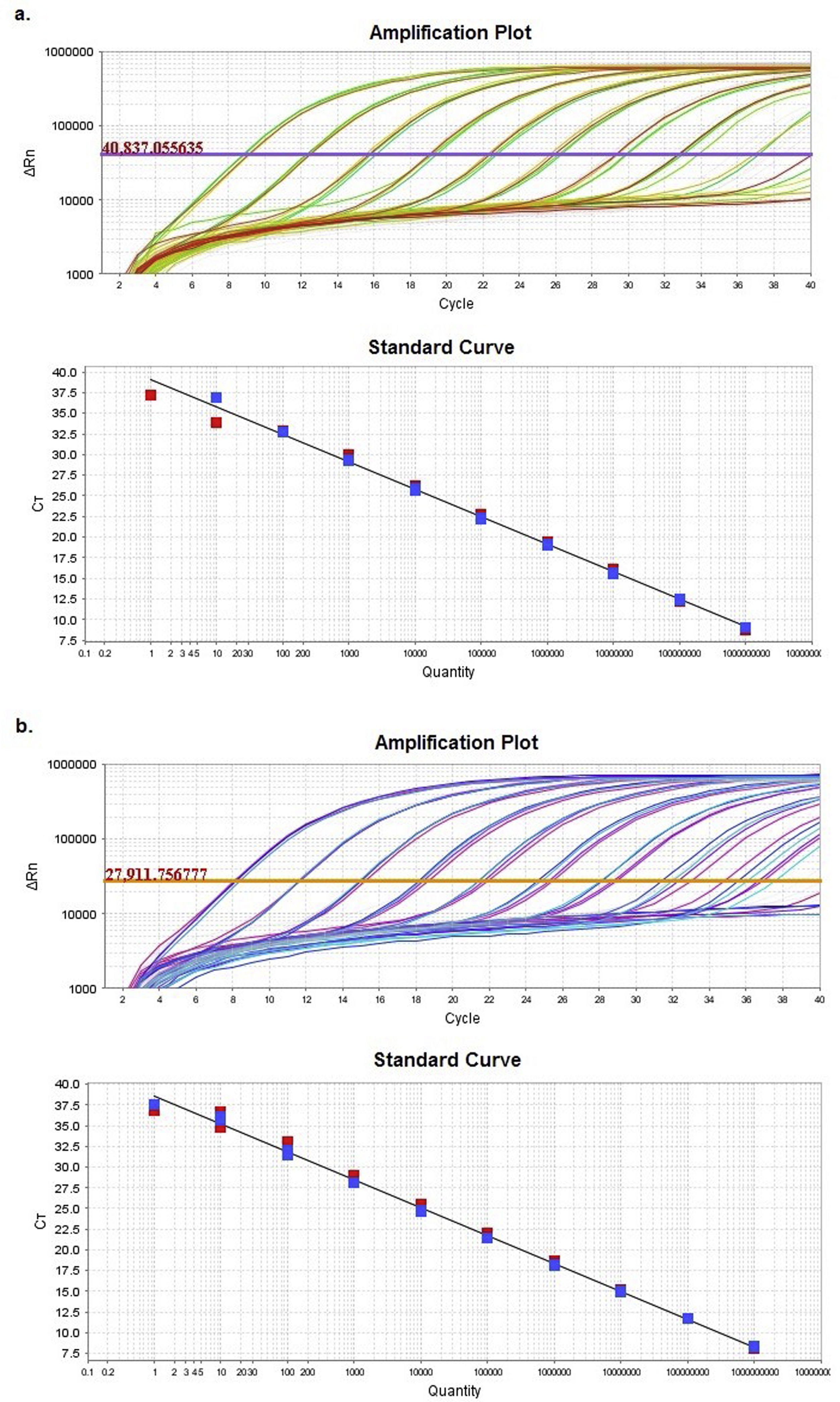
The results were analyzed with High-Resolution Melt Software v3.0.1. (Thermo Fisher Scientific, Waltham, MA, USA). One replica of each clone (H1 to H5) was assigned as “control” for each expected HRMA profile, whereas the other two replicas were kept as “unknown sample”. Additionally, pre-melting and post-melting limits were set around the melting peak to minimize the noise in the reading along the program (Fig. 1).
HRMA repeatability assay
The same previously described controls for haplotypes 1 to 5 (with 104 DNA copies/μl in nuclease-free water) were used following the procedure described for the HRMA haplotype test. PCR conditions are detailed in Suppl. Table 4.
Intra-plate and inter-plate variability were analyzed in this assay. For intra-plate variability, each haplotype was seeded in triplicate in a 96-well plate and this scheme was repeated varying positions in the same plate. Inter-plate variability was assessed by performing and repeating this latter scheme for three consecutive days. Each day was considered a replica.
The results from this assay were analyzed with High-Resolution Melt Software v3.0.1.
The Tm values obtained for each sample of the Z1 and Z2 fragments were analyzed with a nested ANOVA test to calculate the variability between replicates.
Concordance between HRMA and sequencingSixty-four ILTV field samples and two vaccines (Table 1) were previously characterized and their haplotypes were determined by sequencing8. Each DNA sample was seeded in triplicate together with clones H1 to H5 as controls. HRMA was performed on the Z2 fragment following the PCR protocol described above in the HRMA haplotype test. The characterization results obtained by sequencing and HRMA were compared and the concordance between both methodologies was determined using Cohen's κ coefficient6, according to the following formula:
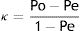
where:
Po=proportion of observed matching between sequencing and HRMA in the establishment of a haplotype. Matching determinations/total of determinations.
Pe=proportion of expected matching occurring by chance.
Characterization of new field strains by HRMAThe setup of all the HRMA protocol parameters was checked by using five selected positive samples for ILTV that were collected between 2013 and 2015 (Table 1). Additionally, a sequencing analysis was performed on PCR amplicons (1279bp) of the US5 gene containing the Z1 and Z2 regions. These PCR fragments were purified using the QIAquick PCR purification kit (Qiagen Inc.), according to the manufacturer's recommendations. A bi-directional DNA sequencing was performed by the Sanger technique (3500xL Genetic Analyzer, Applied BiosystemsTM), by using primers IgJ-Fw and IgJ-Rv13 (Table 3 in supplementary material), and internal primers gJ-ForS622 and gJ-RevS500. Finally, sequences were aligned, edited and analyzed using BioEdit Sequence Alignment Editor Software15.
ResultsComparison of HRM sensitivity using different diluentsAmplification plots and standard curves from the Z1 and Z2 fragments were generated in two different diluents: nuclease-free water and matrix (Suppl. Table 5).
Supplementary table 5 displays slope and regression coefficient (R2) values together with the amplification efficiency (Eff%) obtained for the quantification standard curves by using StepOneTM Software v2.3. In both cases, either efficiency or R2 showed values with an excellent adjustment between the Ct and the numbers of DNA copies.
Multiple linear regression between both diluents showed no significant differences (p<0.05) between the regression lines, as evidenced by the statistical similarity of slopes and intercepts.
Based on the results obtained, the optimal concentration range was between 102 and 109 DNA copies/μl (9.00<Ct<32.5). These concentrations allowed a reliable analysis of the Z1 and Z2 fragments.
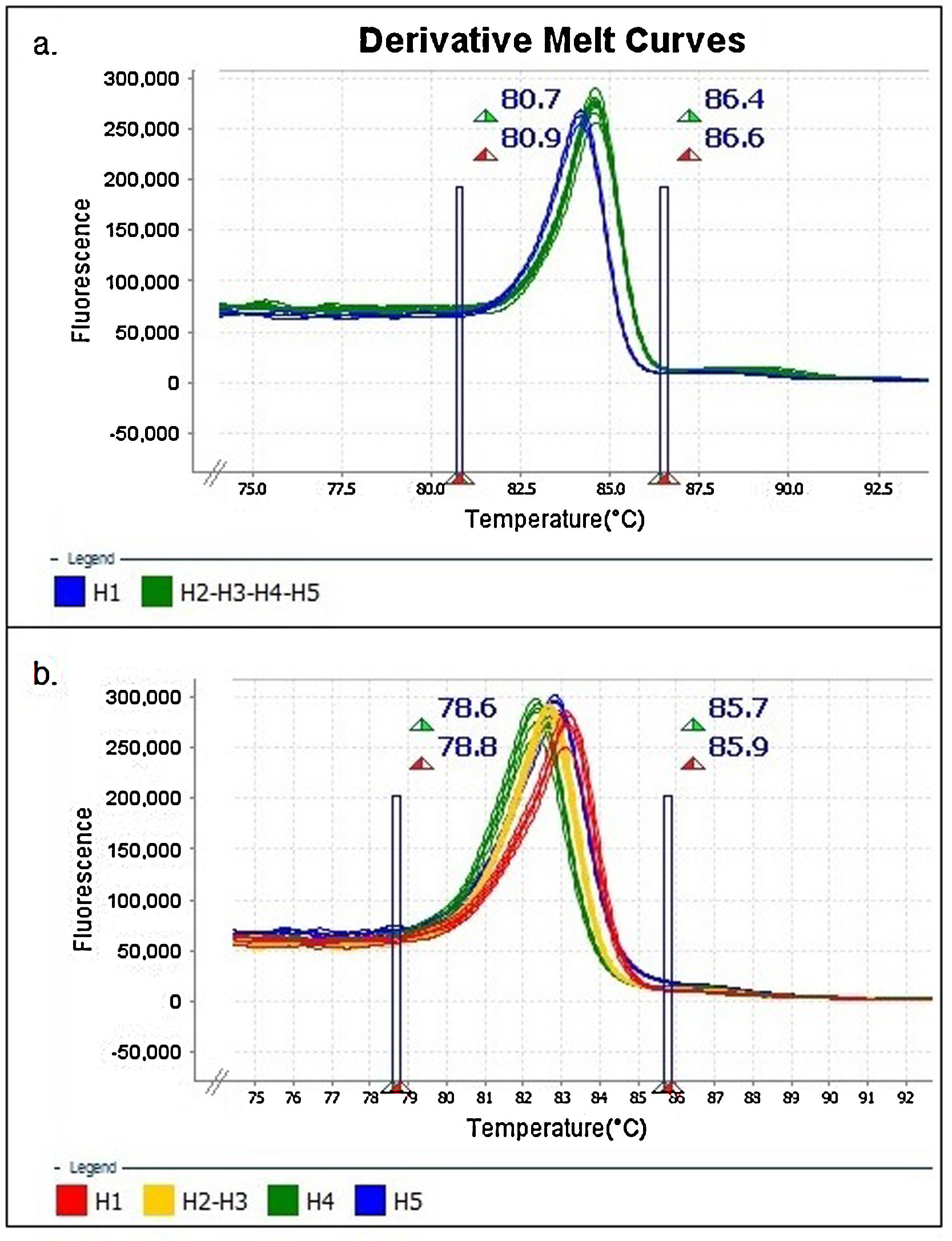
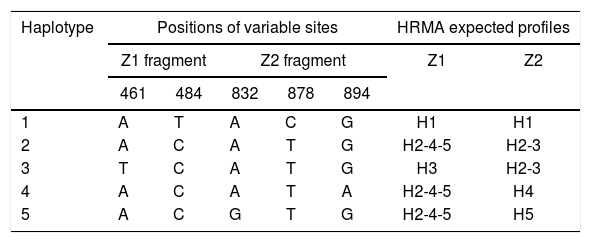
Identification of haplotypes by HRMAAn in silico analysis of the Z1 fragment indicated that there should be three different curve profiles containing the five haplotypes grouped as follows: one curve for haplotypes 2, 4 and 5 (H2-4-5) and the other two for haplotype 1 (H1) and haplotype 3 (H3), respectively. On the other hand, the four expected curve profiles for the Z2 fragment should correspond to H1, H2-3, H4 and H5. Therefore, a proper determination of the haplotype of an ILTV sample needs a combined analysis of the curve profiles of both fragments (Table 2).
Nucleotide base combinations generating each haplotype and HRMA expected profiles.
| Haplotype | Positions of variable sites | HRMA expected profiles | |||||
|---|---|---|---|---|---|---|---|
| Z1 fragment | Z2 fragment | Z1 | Z2 | ||||
| 461 | 484 | 832 | 878 | 894 | |||
| 1 | A | T | A | C | G | H1 | H1 |
| 2 | A | C | A | T | G | H2-4-5 | H2-3 |
| 3 | T | C | A | T | G | H3 | H2-3 |
| 4 | A | C | A | T | A | H2-4-5 | H4 |
| 5 | A | C | G | T | G | H2-4-5 | H5 |
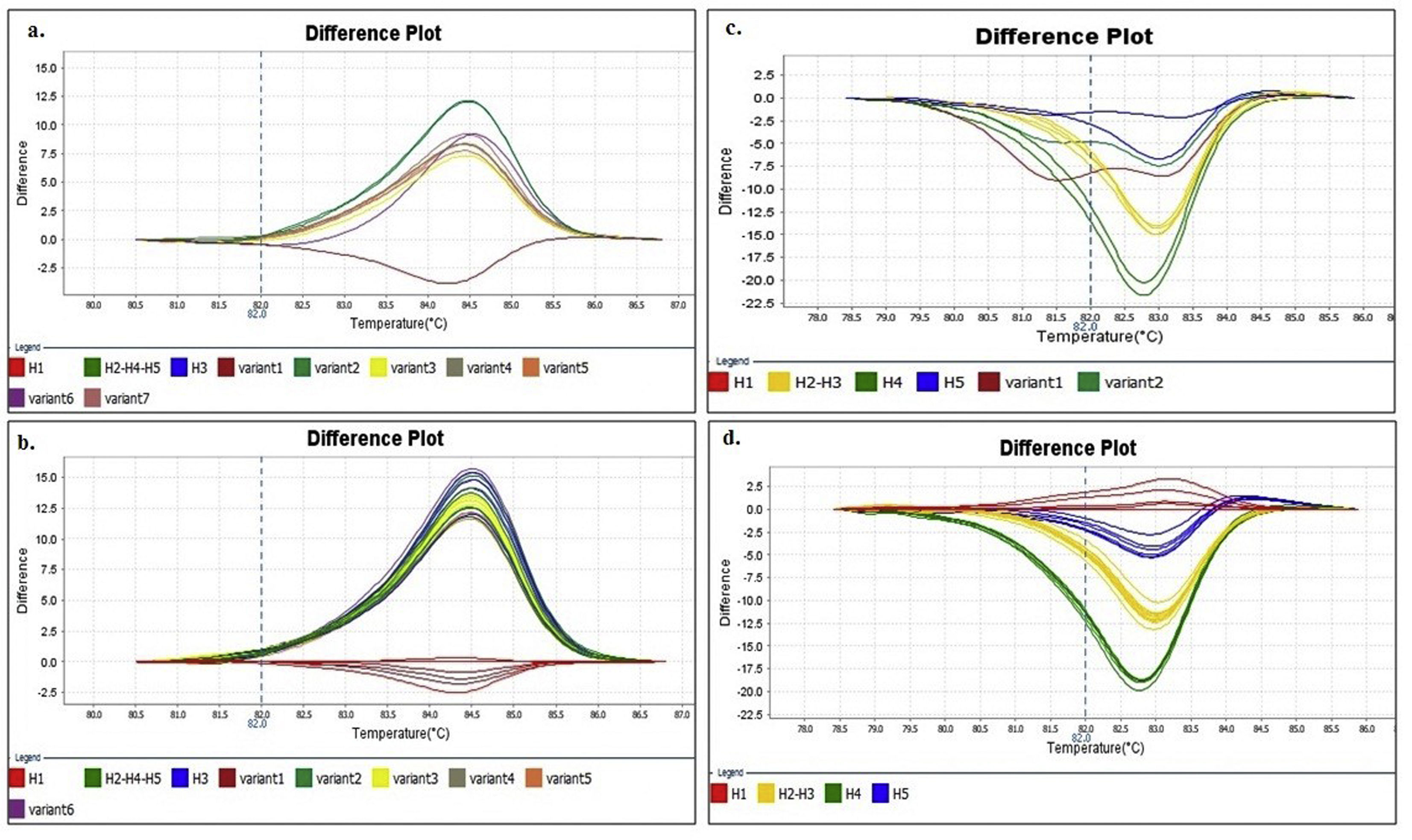
Upon analyzing the Z1 fragment, it was observed that all samples were classified as variants. In the case of the field samples, the assignment of haplotype identity was unsuccessful with this region.
Similar results were obtained when using different concentrations (copies/μl) of DNA template and with the two evaluated dilution media (nuclease-free water and elution buffer) (Figs. 2A and B). Furthermore, as expected, the shape of the curves was irregular in the case of the clones with low concentration (102copies/μl and a Ct>30, Fig. 2A).

Denaturing profiles obtained after amplification. A. Z1 fragment with 102 copies/μl of clones diluted in nuclease-free water and B. 104 copies/μl diluted in nuclease-free water and elution buffer. C. Z2 fragment with 102 copies/μl of clones diluted in nuclease-free water and D. 104 copies/μl diluted in nuclease-free water and elution buffer.
In the Z2 fragment analysis, clones with 104 copies/μl (Ct<30) showed four different curve profiles that clearly distinguished haplotype 1, haplotypes 2 and 3 together, haplotype 4 and haplotype 5, as expected according to the sequence of this fragment (Fig. 2D). The average percentage of confidence of the software in assigning the haplotypes was: 93.3 for H1, 99.0 for H2-3, 99.5 for H4 and 81.4 for H5.
Although the nucleotide sequences did not change in this analysis, a low copy number of clones from this region (102 copies/μl and a Ct>30) reduced the possibility of characterizing the samples and increased the occurrence of discordance in the curve profiles (variants) (Fig. 2C).
Because no additional information could be extracted from the Z1 fragment, any further analysis henceforth only contained the Z2 fragment in order to discriminate between H1, H2-3, H4 and H5.
No differences were detected using water or buffer as diluents in any of the assays.
HRMA repeatability assayIntra-plate as well as inter-plate repeatability assays showed no significant difference between replicates. The tested clones presented the same profile in both assays, with no significant difference of Tm values between replicates. Tm values with nested ANOVA showed a variability coefficient below 0.1.
Concordance between HRMA and sequencingOf the 66 analyzed samples, only one yielded an unexpected haplotype. Its profile showed it as a variant, even though it had been assigned as haplotype 3 by sequencing (Table 1). Further DNA extraction was performed to this sample to improve this result. However, the analysis over this fresh DNA remained inconclusive.
Cohen's κ coefficient was calculated to determine the concordance between HRMA and sequencing. Samples with haplotypes 2 and 3 were grouped together in sequencing results for a fair comparison with HRMA, taking into account that the Z2 fragment analysis exhibited one curve profile for both haplotypes. This κ value was 97.49%, thus indicating a high percentage of coincidence between both methodologies under these conditions.
Characterization of new field strains by HRMAField strains named ER13_14, ER13_21, ER13_22, ER15_37 and ER15_38 were characterized by HRMA (Table 1) to determine the haplotype and to test the conditions that were set up for this HRM analysis. Subsequently, they were sequenced to verify the identity determined by the haplotype profile.
The Z2 fragment analysis by HRMA showed profiles corresponding to haplotype 5 in four samples (ER13_21, ER13_22, ER15_37 and ER15_38) and to H2-H3 in another sample (ER13_14).
The sequence analysis confirmed the identity of all analyzed samples.
DiscussionIn this study, we optimized an HRMA specific for ILTV that would allow a faster and more economical characterization of field strains than the classical sequencing technique. HRMA is simple, economical and accurate and these characteristics make it a valuable tool as a screening method prior to the sequence analysis.
Instead of amplifying the 1279-bp fragment of the US5 gene studied by Craig and collaborators8, we designed primers to amplify shorter regions to ensure maximum sensitivity, in accordance with the recommendation of amplicon size for a better discrimination25.
Some critical points must be taken into consideration when performing HRMA. For instance, the accuracy of the results depends to a large extent on the quality of the real-time amplification25. Those samples that amplify late or fail to reach a high signal plateau in the PCR phase can result in inconclusive or low-resolution HRMA data.
With this fact in mind, we assayed different factors that could possibly affect the quality of the DNA and, hence, the reliability of the results, in order to optimize the technique.
The HRMA performed on samples diluted alternatively in nuclease-free water or matrix showed that the presence of heterologous DNA and other cellular metabolites in the matrix did not interfere with the accuracy of the technique. The absence of both nonspecific amplification and/or alteration of the curve shape in HRMA is essential for the proper characterization of a pathogen.
Relative stabilities of native and denatured DNA may be altered in concentrated salt solutions. This effect is reflected in changes in the Tm of the DNA24 and can result in low sensitivity, poor reproducibility and incorrect genotype calls. In the present study, we evaluated two different eluents that are commonly used in DNA extraction. No evident differences were observed between the curve behavior using nuclease-free water and the elution buffer (provided by the DNA extraction kit).
Another factor to consider is the concentration of the DNA template in the sample. There are different suggestions about the cutoff of the Ct that results in reliable curve profiles: White and Potts30 recommend employing this tool in those cases where the Ct value is below 35; however, the equipment manufacturer suggests using it with Ct values below 30. The influence of this parameter was assessed by analyzing two different concentrations (102 and 104 DNA copies/μl) of the haplotype clones, the obtained Ct values being 32.5 and 26.0, respectively.
At the highest clone concentration (Ct≅26.0), the results of the analysis of the Z2 fragment yielded well defined curve profiles that were clearly distinguishable from each other. The profiles obtained at a lower clone concentration (Ct≅32.5), however, showed more irregular curves. These altered curve profiles have led to classify these clones as variants, therefore, hindering the characterization of the samples into their corresponding haplotypes. Based on this result, HRMA would be reliable only to identify samples with a Ct value below 30.
Unexpectedly, an experimental limitation was evident during the adjustment of the technique with regard to the discrimination between the nucleotide transversion A/T at position 461 in the Z1 fragment. This position is responsible for differentiating haplotype 3 from 2, 4 and 5. Each transition and transversion has different variations in their Tm; the change A/T has the lowest variation in Tm, with a deviation lower than 0.25°C17. Although the equipment used in this study makes fluorescence readings every 0.1°C, we were unable to discriminate H3 from the other haplotypes within the Z1 fragment. In a comparison study of nine instruments capable of performing HRMA, the researchers reported certain variability between the predicted Tm value for a specific mutation and the measured one17.
Another reason that could explain this limitation is the position of the mutation within the genomic region. Indeed, curve profiles could differ depending on the surrounding sequences of a nucleotide variant.
Sequencing is generally regarded as the gold standard for assessing sequence variation with an accuracy approaching 99.9%26 and therefore the assessment of the concordance value between both techniques is essential. In terms of the studied genomic region (US5), and under the conditions used in this study, the concordance (97.49%) between HRMA and sequencing was high.
These results support the idea that HRMA can be applied as an effective screening tool for ILTV strains. Furthermore, although the analysis of the Z1 fragment was dismissed because of the impossibility to differentiate H2 from H3, the Z2 fragment analysis allowed us to distinguish between the other haplotypes and showed a very good concordance with sequencing. Taking these results into account, the use of sequencing for the characterization of some haplotypes (H1, H4 and H5) would not be necessary, although the differentiation of H2 and H3 would still require this standard method. Sequencing would also be essential to give a new identity in the event of the emergence of a new variant.
Altogether, the analysis of the Z2 fragment allowed us to identify haplotypes H1, H4 and H5 and therefore to reduce the number of samples to be sequenced.
Although in this study we used HRMA only to characterize field samples, we do not dismiss the possibility of using this technique as a diagnostic tool as well, because a wide range of DNA concentrations (102 to 109 DNA copies/μl) generate a proper amplification signal. To achieve this, firstly it will be necessary to determine if the Z2 fragment of US5 is specific enough to make the diagnosis by comparing the results with the validated protocol for ILT detection2. This procedure would give us the advantage of detecting and characterizing some samples in just one step. In case of H2/H3, the sample could be purified and sequenced in a step forward. Therefore, as previously underlined by others26,31, this procedure would be an available scanning technique that can be performed in the same container used for PCR amplification, consequently preventing possible environmental contamination.
In summary, HRMA showed its potential to replace sequencing for ILTV characterization. Although in this case it was not possible to differentiate H2 from H3, this tool showed a very good correlation to sequencing, thus providing the possibility of reducing workflow and the number of field samples to be sequenced.
FundingFunding for this study was provided by PNBIO-1131032 from the Instituto Nacional de Tecnología Agropecuaria (INTA) and PICT-12-0476 from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT).
Conflict of interestThe authors declare that they have no conflicts of interest.
The authors would like to thank Dr. Fondevila for his assistance with the statistical analysis of the results.