


Los agujeros parietales son pequeñas formaciones ubicadas en la calota craneana sobre los huesos parietales, en topografía parasagital. Estos orificios miden en promedio 1mm, por lo que si superan los 5mm, se denominan agujeros parietales gigantes (APG). Se asocian a distintos tipos de craneosinostosis, displasias corticales, microcefalia, defectos oculares o del oído, sindactilia y polidactilia con hipoplasia distal de la clavícula y ausencia del acromion, retraso mental, exostosis múltiples, disostosis craneofacial y alteraciones de los vasos intracraneales1,2. Muy rara vez se ha citado su vínculo con malformaciones cardíacas3.
Presentamos el caso de una niña de 3 meses de vida, que había nacido pretérmino (31 semanas) con un peso 1470 gramos. Al nacer, había requerido reanimación cardiopulmonar y tuvo una internación de 63 días en Neonatología por una comunicación interventricular (CIV) perimembranosa moderada y una comunicación interauricular (CIA) tipo ostium secundum. Fue derivada a nuestra institución para el tratamiento de su malformación cardíaca.
Al ingreso la paciente se encontraba sin fiebre, irritable y con distrés respiratorio. Las fontanelas anterior y posterior eran normales y no presentaban tensión. Sin embargo, detrás de la fontanela anterior se observó una región de partes blandas sobreelevada y de morfología ovalada, que a la palpación se revelaba renitente y con un borde óseo que la delimitaba. Dados los antecedentes cardíacos y los hallazgos en la calota craneana, se solicitó una ecografía transfontanelar (ETF) y una tomografía computada (TC) para descartar alteraciones sindrómicas. En nuestra institución no se llevó a cabo radiografía de cráneo.
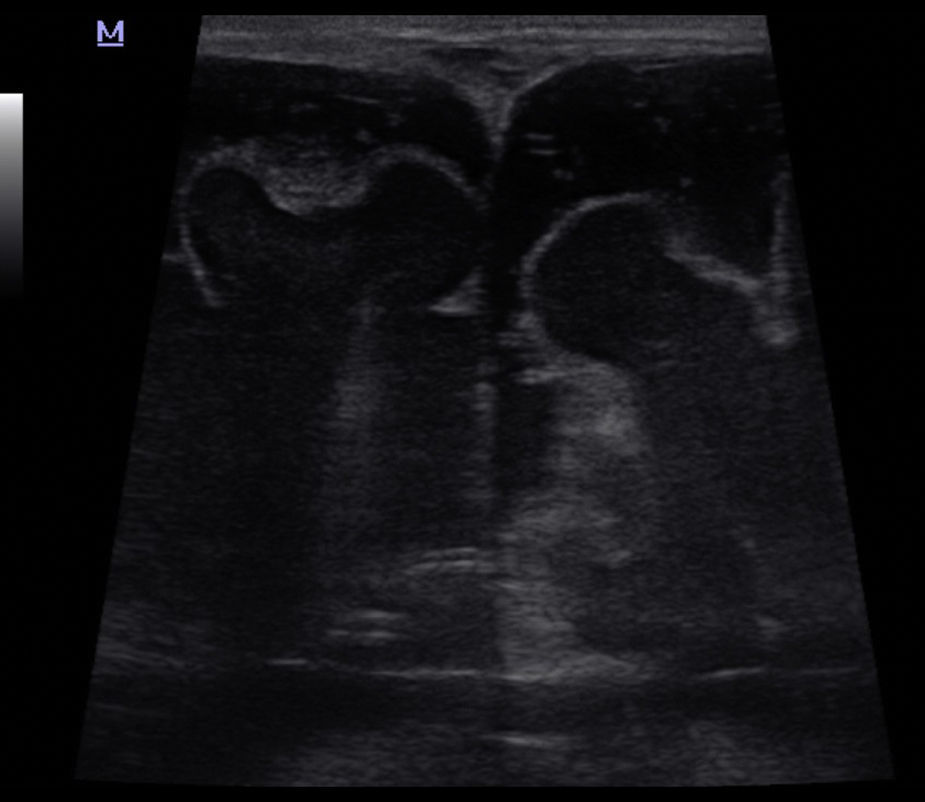
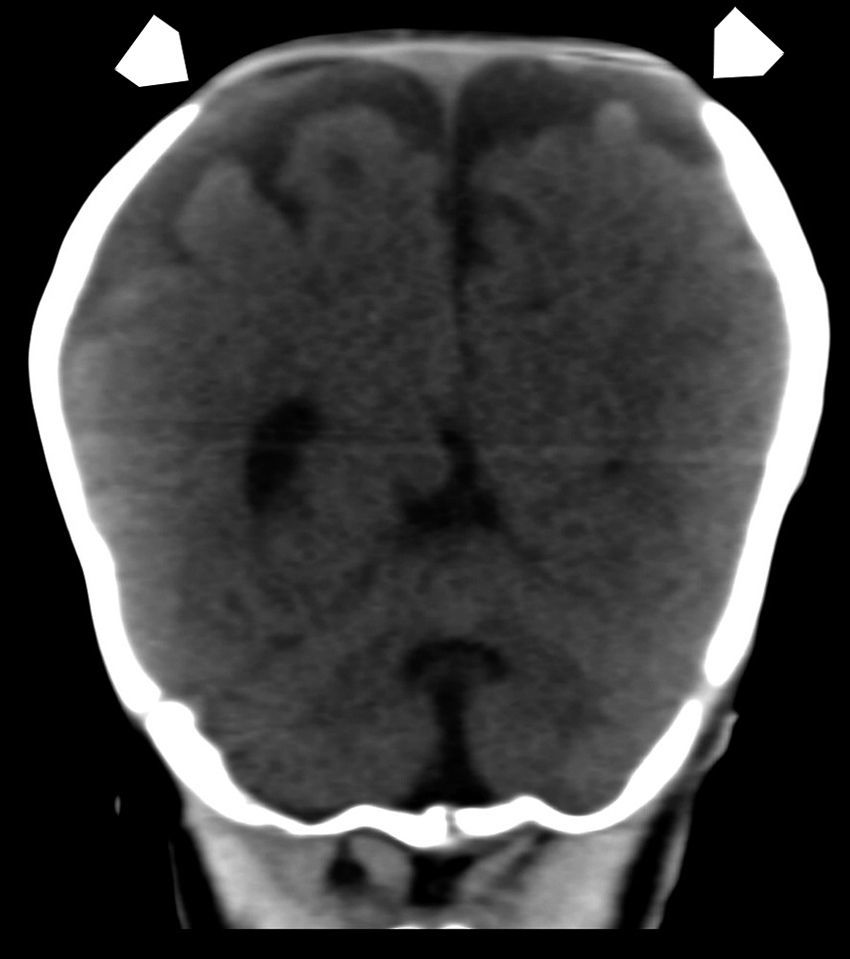
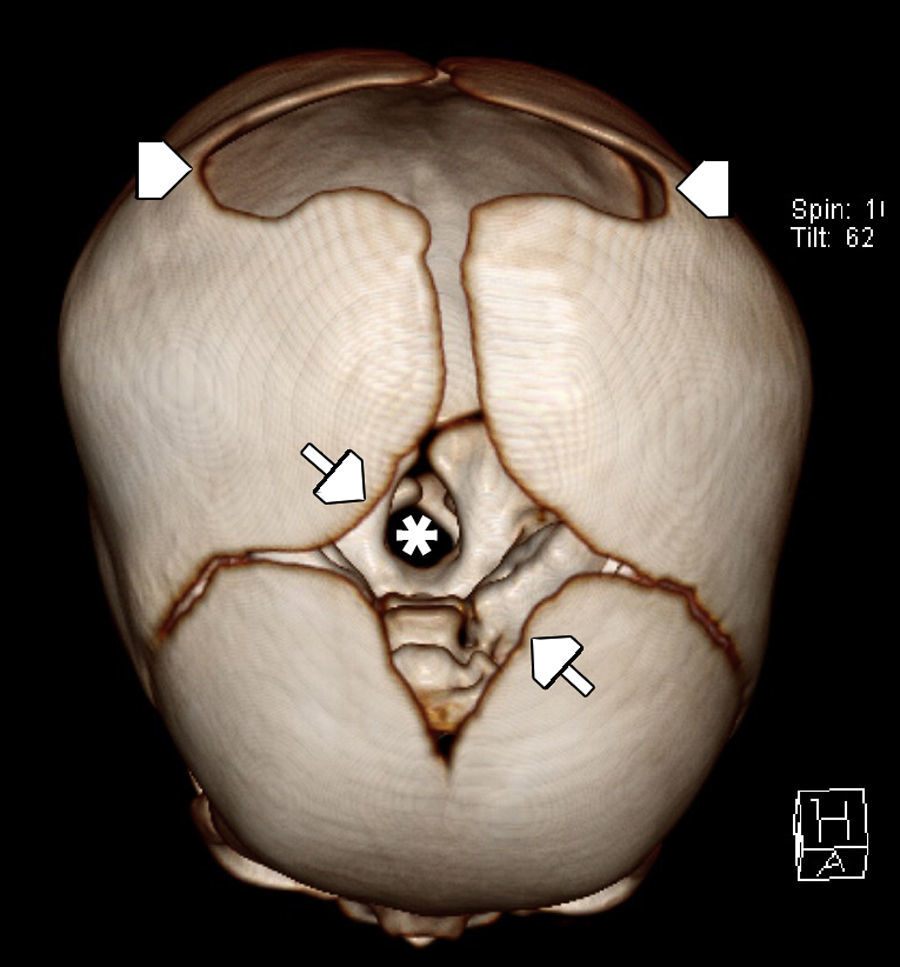
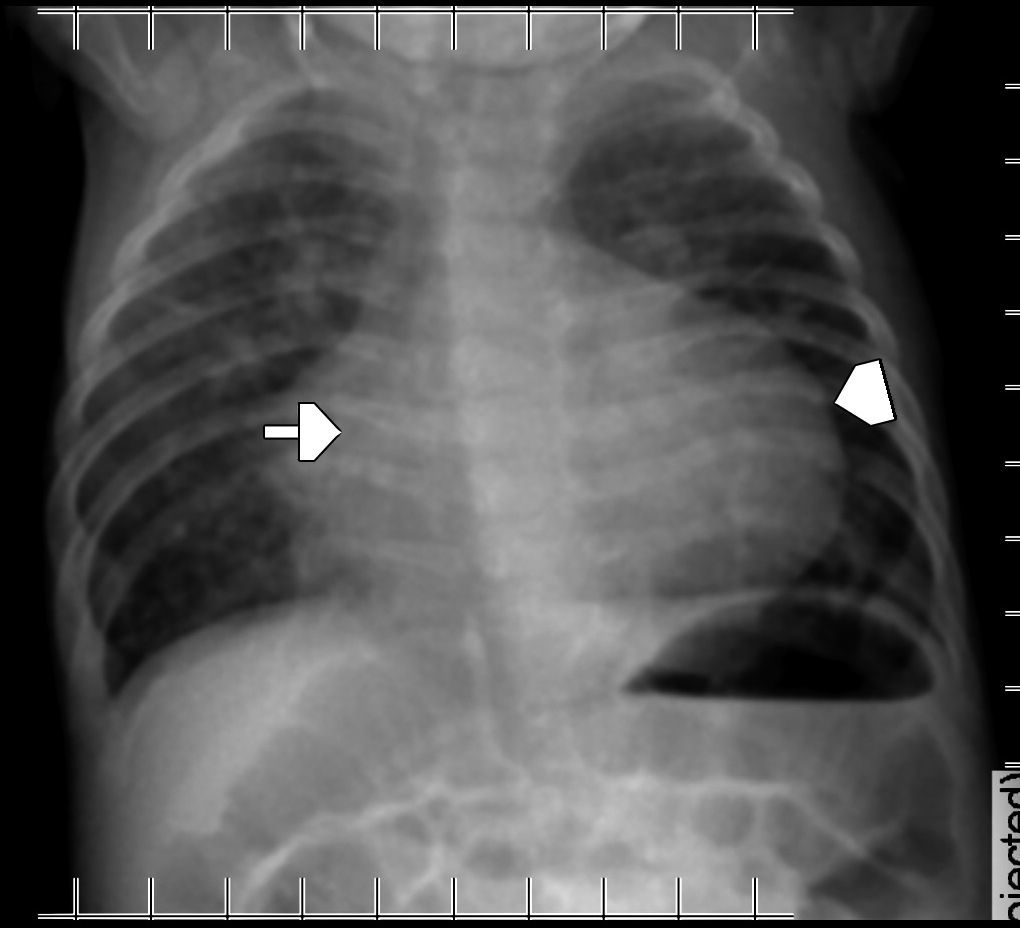
La ETF evidenció un extenso defecto óseo con una leve protrusión meníngea y del espacio aracnoideo a través de la diastasis ósea parietal, sin otras alteraciones encefálicas (fig. 1). Por su parte, la TC advirtió mejor el extenso defecto en la conformación de ambos huesos parietales (fig. 2), con una gran solución de continuidad y una amplia zona sin revestimiento óseo a nivel de la calota craneana, que creaba una “pseudofontanela posterior” (fig. 3) por donde protruían el espacio aracnoideo y las meninges, un hallazgo compatible con APG. No se observaron otras alteraciones de las estructuras extra e intracraneales, pero en la radiografía de tórax se apreció la típica imagen de corazón en bota de las cardiopatías (fig. 4). La paciente fue tratada por su patología cardíaca y dada de alta para control en su localidad.

 donde se evidencia una leve protrusión del espacio aracnoideo y de las meninges.")
 a través de la fontanela anterior (flechas) y, por detrás de ella, una deformidad de ambos huesos parietales, creando una pseudofontanela llamada agujero parietal gigante (cabezas de flecha).")
Tomografía computada de cráneo, en reconstrucción tridimensional, en vista superior, en la que se observa el agujero magno (asterisco) a través de la fontanela anterior (flechas) y, por detrás de ella, una deformidad de ambos huesos parietales, creando una pseudofontanela llamada agujero parietal gigante (cabezas de flecha).
 y el ventrículo derechos (cabeza de flecha), configurando el corazón en bota.")
El hueso parietal presenta, en porcentaje variado, pequeños orificios que pueden ser únicos, en par (uno a cada lado), triples (dos de un lado y uno del otro) o pares a cada lado de la sutura sagital, cercanos al Lambda y conocidos como forámenes o agujeros parietales4. Estos permiten el pasaje de venas emisarias que comunican con el seno longitudinal superior. La frecuencia del foramen parietal bilateral alcanza el 59% y el diámetro promedio oscila entre los 0,6 y 1,5 mm 5. Si su diámetro excede los 5mm, se llaman agujeros parietales gigantes o foramina parietalia permagna. Estos no constituyen simplemente un agujero parietal aumentado de tamaño, sino una patología benigna de defecto en la osificación6.
El tratamiento quirúrgico puede ser necesario cuando el diámetro es muy grande o persiste aumentado de tamaño con el crecimiento del niño. En ambos casos se busca, por medio de injertos óseos autólogos o mallas, proteger el cerebro subyacente7. Los agujeros suelen tener formas variadas, pudiendo ser ovales, redondos, rectangulares, irregulares o con depresiones. La multiplicidad de tamaños, la ausencia de manifestaciones clínicas y la falta de asociación sindrómica conduce a un diagnóstico tardío.
La entidad también es conocida como Catlin mark. Su nombre se debe al apellido de la familia en la que Goldsmith8 describió por primera vez la presencia de esta alteración en el cráneo de varios integrantes del mismo grupo familiar, demostrando así su relación hereditaria. Estudios posteriores han confirmado su herencia autosómica dominante debida a mutaciones heterocigotas en el gen MSX29,10.
Los APG constituyen una condición benigna que puede estar asociada a múltiples alteraciones morfológicas o genéticas. Conocer su clínica y características radiológicas resulta relevante, ya que ante este hallazgo hay que estudiar ampliamente al paciente y su familia.
Atentamente, los autores
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
