







El síndrome de Rett (SR) es un trastorno del neurodesarrollo que afecta casi exclusivamente a niñas y cursa secundariamente con autismo. Es poco frecuente y consta de 5 formas clínicas, una clásica y el resto atípicas que comprometen de manera general la habilidad manual, el lenguaje y la motricidad amplia unida a la aparición de estereotipias y epilepsia precoz. Con el objetivo de actualizar la información sobre SR, se aplicaron los descriptores de búsqueda Síndrome de Rett, genes y «Síndrome de Rett», «Rett Syndrome gene», «Rett Syndrome», «Rett Syndrome gene therapy» y «Rett Syndrome review». Se investigó en los archivos digitales PubMed, Hinari, SCIELO y Medline, y se consultaron los sitios web OMIM, ORPHANET, GeneMap, Genetests, Proteins y Gene, entre otros. Entre 1.348 artículos se seleccionaron 42, los cuales reportan 3 genes causantes del síndrome: MECP2, CDKL5y FOXG. El gen MECP2 está mutado en el 80% de los pacientes con SR clásico así como en el 40% de los afectados con alguna de sus formas atípicas. El SR con epilepsia precoz y la variante congénita se deben fundamentalmente a variaciones en los genes CDKL5 y FOXG1 respectivamente.
ConclusionesEl diagnóstico del SR se basa en criterios clínicos, sin embargo, los avances en la biología molecular y en la genética en particular han abierto el abanico de posibilidades diagnósticas a las diferentes formas clínicas que antes quedaban sin clasificar, a la vez que el análisis molecular permite confirmar el criterio clínico y aportar información en cuanto al pronóstico del paciente.
Rett syndrome (RS) is a neurodevelopmental disorder that exclusively affects girls, and occurs along with autism. It is very uncommon, and has five distinct forms, one classic and the others atypical, which generally compromise manual skills, language, and mobility, and widely associated with the appearance of stereotypy and early epilepsy. With the aim of updating the information about RS, a search was performed in the computer data bases of PubMed, Hinari, SCIELO and Medline, as well as consulting other web sites including OMIM, ORPHANET, GeneMap, Genetests, Proteins and Gene, using the descriptors “Síndrome de Rett”, “genes y Síndrome de Rett”, “Rett Syndrome gene”, “Rett Syndrome”, “Rett Syndrome gene therapy”, and “Rett Syndrome review”. Of the 1,348 articles found, 42 articles were selected, which reported 3 genes causing the syndrome: MECP2, CDKL5 and FOXG. The MECP2 gene is mutated in 80% of patients with classic RS, as well as in 40% of those affected by any of its atypical forms. RS with early epilepsy and the congenital variant are mainly due to variations in the CDKL5 and FOXG1 genes, respectively.
ConclusionsThe diagnosis of RS is based on clinical criteria. However, the advances in molecular biology and genetics have opened a wide range of possibilities for diagnosing the different clinical forms that could not be classified before. Molecular analysis can help confirm the clinical criteria and provided information as regards the prognosis of the patient.
El síndrome de Rett (SR), descrito en 1966 por Andreas Rett1 y definido clínicamente por Bengt Hagberg en 19832, es un trastorno del neurodesarrollo que afecta casi exclusivamente a niñas y cursa secundariamente con autismo. Su prevalencia no es bien conocida, pero se estima entre 0,5 y 1 de cada 10.000 recién nacidos3. Existe una forma clásica caracterizada por un período de normalidad seguido de pérdida parcial de la habilidad manual, el lenguaje y la motricidad amplia y aparición de estereotipias, y 4 formas atípicas: regresión tardía, lenguaje conservado, epilepsia precoz y variante congénita3.
Tras la identificación del gen MECP2 en 1999 por Amir et al.4 fue posible la confirmación molecular de un gran número de pacientes con diagnóstico clínico de SR. Sin embargo, un 20% de las pacientes con SR clásico y un 60% con variantes atípicas5,6 no presentan mutaciones en este gen. La caracterización en 2005 del gen CDKL5, responsable de la mayoría de los casos con epilepsia precoz de este síndrome6, ha permitido el diagnóstico genético en un 3-10% de las pacientes sin diagnóstico molecular previo. Desde 2008, un nuevo gen, el FOXG1, se perfiló como responsable de la variante congénita del SR5,7–10.
El diagnóstico de SR, tanto en su forma clásica como en sus variantes atípicas, se basa en los criterios clínicos, pero el diagnóstico molecular permite confirmar el mismo y aportar información en cuanto al pronóstico. La colaboración entre clínicos y genetistas facilitará la definición de un fenotipo característico de las distintas mutaciones3. Los criterios de exclusión incluyen otros síndromes que causen disfunción neurológica o retraso del desarrollo psicomotor en los primeros 6 meses de vida aunque también hay que tener presente las diferencias fenotípicas que presentan las variantes atípicas del SR11. Con el objetivo de resumir el estado actual del conocimiento acerca de las bases moleculares de este síndrome se realizó la presente revisión.
MetodologíaSe realizó una búsqueda sobre el estado actual del SR, con especial énfasis en las bases genéticas del mismo. Se consultó bibliografías de alto impacto y reciente publicación. Se hicieron búsquedas a través de PubMed, Hinari, SCIELO, Medline y se consultaron páginas dedicadas a estos temas como OMIM, ORPHANET, GeneMap, Genetests, Proteins, y Gene, entre otros.
Los descriptores de búsqueda principales fueron: Síndrome de Rett, genes y Síndrome de Rett, Rett Syndrome gene, Rett Syndrome, Rett Syndrome gene therapy y Rett Syndrome review. Fueron encontrados 1.348 artículos relacionados con los descriptores utilizados; se restringió la búsqueda a los últimos 5 años lo cual redujo el número de artículos. Finalmente se tomaron aquellos reportes con aspectos novedosos sobre los diferentes genes y las proteínas involucradas, sus mutaciones y consideraciones sobre eventuales tratamientos como la terapia génica. De ellos se seleccionaron 42 publicados del 2010 al 2013.
Otros descriptores de búsqueda fueron los genes involucrados específicamente (MECP2, CDKL5, FOXG1) consultados principalmente en los sitios OMIM, ORPHANET, GeneMap, Genetests, Gene y RettBASE: IRSF MECP2 Variation Database de la International Rett Syndrome Foundation. Estas últimas consultadas para la caracterización de los genes, sus principales mutaciones y las proteínas con sus características.
Formas clínicas, variaciones del defecto y sus causasSíndrome de Rett clásicoEl cuadro clínico cursa con un período prenatal y perinatal normal (exceptuando la variante congénita), y un desarrollo normal o casi normal durante los primeros meses de vida. Tras este período, entre los 3 meses y 3 años de vida se pierden las capacidades manuales propositivas, uno de los elementos característicos de la enfermedad, y se produce una regresión de las funciones psicomotoras y de la comunicación. El contacto ocular está muy limitado al igual que en la mayoría de los pacientes con autismo12,13. Aunque es normal al nacimiento, el crecimiento del perímetro craneal sufre un descenso paulatino (que puede conducir a microcefalia) y que puede ser tan precoz como en los 3 primeros meses de vida, constituyendo a veces el primer signo del SR.
Entre el primer y tercer año de edad aparece la seña de identidad del SR: los movimientos estereotipados de las manos (característicamente de lavado, manos juntas, pero también de palmoteo o de aplausos, manos separadas). Pueden aparecer otras estereotipias tales como temblores, caídas bruscas, detención del movimiento, episodios de risa o gritos inmotivados. La marcha suele ser normal al inicio pero se va volviendo amplia y errática; es frecuente el balanceo de un lado a otro12.
Algunos pacientes presentan desde el inicio de la enfermedad alteraciones neurológicas evidentes tales como ataxia del tronco, escoliosis neurógena, crisis convulsivas, alteraciones de la respiración, alteraciones gastrointestinales, disfunción piramidal, hipoacusia neurosensorial leve y neuropatía periférica, pero habitualmente dichos signos se presentan posteriormente de forma progresiva predominando en los primeros estadios de la enfermedad un trastorno generalizado del desarrollo capaz de simular un autismo13.
En esta enfermedad la invalidez es importante, y la falta de cuidados lleva característicamente a estos pacientes a la escoliosis deformante y a la desnutrición grave, por lo que la expectativa de vida está disminuida a pesar de que existen raros casos que han sobrepasado los 60 años de edad12.
Los individuos que cumplen con la mayoría de los criterios diagnósticos de esta enfermedad son clasificados como pacientes con SR clásico. No obstante, el fenotipo es más variado de lo que se describió al principio ya que se han reportado 4 formas atípicas de la enfermedad3. La mayoría de estas variantes son, comparadas con la forma clásica, más leves, especialmente en el grado de disfunción motora12, y se asocian con mucha menor frecuencia que la forma clásica a mutaciones del gen MECP2 ya que se han descrito al menos otros 2 genes implicados: CDKL5 y FOXG13.
Síndrome de Rett con regresión tardíaLa regresión del desarrollo psicomotor se manifiesta tardíamente, generalmente después de los 4 años de edad, y de forma más insidiosa.
Síndrome de Rett con lenguaje conservadoTras la fase de regresión los pacientes consiguen pronunciar algunas palabras que fueron aprendidas antes de manifestarse la enfermedad (nunca adquiridas después de la fase de regresión).
Síndrome de Rett con epilepsia precozLos pacientes cumplen con los criterios de la forma clásica pero los síntomas comienzan antes de los 6 meses de edad y la presentación clínica inicial está dominada por las crisis epilépticas lo cual enmascara el diagnóstico.
Variante congénita del síndrome de RettNo existe un período de normalidad en el desarrollo psicomotor, pero se cumplen los criterios del SR clásico. Cada vez se describen más casos12.
EtiologíaEl SR clásico, así como las variantes con regresión tardía y lenguaje conservado son consecuencia, mayoritariamente, de mutaciones en el gen MECP2 con herencia dominante ligada al cromosoma X3. Por otra parte, el SR con epilepsia precoz y la variante congénita se deben, fundamentalmente, a variaciones en los genes CDKL53 y FOXG13 respectivamente.
MECP2LocusEl gen MECP2 está mutado en el 80% de los pacientes con SR clásico así como en el 40% de los afectados con alguna de sus formas atípicas y se encuentra ubicado en el cromosoma X (Xq28)3,14. El gen contiene 4 exones14 (fig. 1).
Acción del gen
Codifica la proteína 2 de unión de la metil-citosina-guanosina (MECP2)1–14, una proteína nuclear altamente conservada15.
Producto génicoLa proteína codificada tiene 485 aminoácidos y 53kDa de peso molecular14 y se expresa en todo el organismo pero con más intensidad en el cerebro12. Tiene 4 dominios: uno altamente conservado de unión a CpG metilados (MDB) (en el genoma, la mayoría de los residuos de citosina en los dinucleótidos CpG están metilados) que consta de 85 aminoácidos, otro de represión de la transcripción (TRD), uno de señalización de localización nuclear (NLS) y un dominio de segmento carboxilo terminal (CTS)14.
Existen 2 isoformas, A y B, que difieren en las secuencias de sus extremos aminos terminales y en los sitios de inicio de la transcripción: la isoforma A es codificada por los exones 2, 3 y 4, y la B por el 1, 3 y 414,16.
La MECP2 tiene función reguladora ya que se une a regiones metiladas en el ADN, independientemente de la secuencia, y recluta represores y proteínas remodeladoras de la cromatina como HDAC tipos 1 y 2 y las metiltransferasas H3K9 y DNMT1 que forman el complejo correpresor SIN3A. Este complejo es responsable de la compactación de la cromatina y de la supresión de la transcripción génica de los promotores sometidos a su control15,17. Sin embargo, el mapeo reciente de los sitios de unión al ADN de la proteína que nos ocupa ha mostrado que la mayoría de los promotores a los que se une no están metilados y permanecen activos lo cual indica que pueda tener otras funciones además del silenciamiento de la transcripción. Asimismo, esta proteína también se une a regiones específicas en los intrones y esto indica que puede representar un papel fundamental en el splicing o corte y empalme del ARN mensajero18.
La expresión de la MECP2 es especialmente alta en el cerebro y sus niveles cambian durante el desarrollo del sistema nervioso: son altos en la etapa embrionaria, bajos al nacimiento y luego comienzan a aumentar nuevamente15. Durante la infancia se observa un incremento de las neuronas corticales positivas para esta proteína, mientras que las estructuras más profundas muestran un porcentaje constante de neuronas positivas para ella desde las 35 semanas de edad gestacional. De esta forma, las regiones neuronales generadas de forma temprana y, por lo tanto, más maduras, expresan más intensamente la proteína durante el período posnatal inmediato. Los cambios más significativos en la expresión se producen en regiones que maduran después (como la corteza, el cerebelo o el hipocampo), correlacionándose dicho aumento en la expresión con la sinaptogénesis de estas regiones12.
La MECP2 es fundamental para el mantenimiento y modulación de las sinapsis y la complejidad de las dendritas15, y es de vital importancia que esté sometida a una estricta regulación ya que a través de animales de laboratorio se ha observado enfermedad grave tanto por exceso como por defecto en la producción de dicha proteína y a edades diferentes19. Stuss et al.20 demostraron que, en ratones, la ausencia de esta proteína no afectaba significativamente la estructura cerebral pero sí los procesos de maduración neuronal y sinaptogénesis al observarse alteraciones especificas en la morfología y función de las neuronas como, por ejemplo, una reducción en la ramificación de las dendritas15. En otro estudio se comprobó que la sobreexpresión de la proteína, también en ratones, producía un aumento de la complejidad dendrítica y de la longitud de los axones en las neuronas corticales, de lo que se ha deducido que dicha proteína regula directamente la maduración neuronal y la sinaptogénesis e influye en las elongaciones dendríticas y los procesos de ramificación21. Nguyen et al.22 han planteado que estas alteraciones morfológicas son consecuencia de la reducción postranscripcional de proteínas cruciales para la función sináptica como los receptores de las cinasas CaMKII α/β, AMPA y la N-metil-d-aspartato y otras proteínas como la sinapsina18.
Estudios recientes han revelado que la pérdida de función de la MECP2 en ratones conduce a una reducción notable de las sinapsis en las neuronas del hipocampo. Notablemente, la reactivación de la proteína revierte el fenotipo patológico lo cual indica que el daño neuronal producto de alteraciones en la proteína puede ser reparado y esto, por supuesto, tiene importantes implicaciones terapéuticas18.
Tipos de mutaciones y consecuencia de su expresiónMayormente las mutaciones son de novo por lo que el SR se presenta de forma esporádica en el 99% de los casos y el riesgo de la pareja de tener otro hijo afectado es inferior al 1%13.
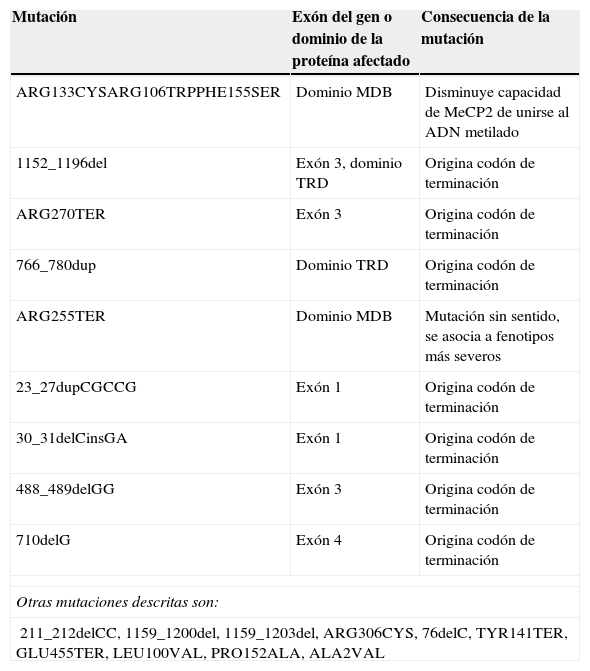
Se han identificado más de 225 mutaciones en el gen MECP2 aunque se encuentran solo 8 en el 70% de los pacientes con SR que cumplen completamente los criterios de consenso12 (tabla 1). La gran mayoría de las mutaciones representan cambios de un único nucleótido pero, entre las mutaciones específicas, las deleciones e inserciones a pequeña escala representan el 60% de las mismas, las primeras agrupadas en la región C-terminal de MECP212.
Algunas mutaciones en el gen MECP2 y la proteína MeCP2 y las consecuencias de su expresión
| Mutación | Exón del gen o dominio de la proteína afectado | Consecuencia de la mutación |
|---|---|---|
| ARG133CYSARG106TRPPHE155SER | Dominio MDB | Disminuye capacidad de MeCP2 de unirse al ADN metilado |
| 1152_1196del | Exón 3, dominio TRD | Origina codón de terminación |
| ARG270TER | Exón 3 | Origina codón de terminación |
| 766_780dup | Dominio TRD | Origina codón de terminación |
| ARG255TER | Dominio MDB | Mutación sin sentido, se asocia a fenotipos más severos |
| 23_27dupCGCCG | Exón 1 | Origina codón de terminación |
| 30_31delCinsGA | Exón 1 | Origina codón de terminación |
| 488_489delGG | Exón 3 | Origina codón de terminación |
| 710delG | Exón 4 | Origina codón de terminación |
| Otras mutaciones descritas son: | ||
| 211_212delCC, 1159_1200del, 1159_1203del, ARG306CYS, 76delC, TYR141TER, GLU455TER, LEU100VAL, PRO152ALA, ALA2VAL | ||
Fuente: OMIM On Line Mendellian Inheritance in Man14.
Los signos y síntomas del SR son dependientes de la edad del paciente y se piensa que las diferentes mutaciones del gen MECP2 producen una sinaptogénesis anormal en distintas etapas del desarrollo, es decir, que se produciría una alteración de la formación y maduración de las sinapsis debido a la imposibilidad por parte de una MECP2 mutada de poder suprimir la expresión de genes que deberían ser silenciados durante este proceso. Aunque la presencia de esta proteína no requiere la formación de sinapsis, la cantidad se incrementa conforme aumenta la formación de sinapsis. Las neuronas maduras que han formado sinapsis inducen niveles plenos de la proteína y en animales de experimentación se ha comprobado que posee una función biológica crítica en las etapas finales del desarrollo neuronal12.
Varones con síndrome de Rett producto de mutaciones en MECP2Inicialmente se pensaba que el SR al ser una enfermedad con herencia dominante ligada al cromosoma X tenía letalidad en varones, pero con el descubrimiento de los diferentes genes involucrados en dicha enfermedad y la posibilidad de un diagnóstico molecular para estos pacientes se comprobó la presencia de niños afectados. El fenotipo de estos niños afectados es extremadamente variable y usualmente más severo que en las niñas con SR14.
Los varones con mutaciones en MECP2 se clasifican en 4 grupos14:
- •
Varones con cariotipo 47,XXY.
- •
Varones con cariotipo 46,XY con mutaciones en MECP2 que causarían SR clásico en niñas pero que en ellos provocan la variante congénita y muerte temprana.
- •
Varones con cariotipo 46,XY con mutaciones en MECP2 que no han sido reportadas en niñas.
- •
Varones con cariotipo 46,XY con niveles extremadamente elevados de la proteína MECP2 debido a duplicaciones.
El fenotipo de los pacientes con mutaciones en el gen MECP2 depende de 2 factores: el patrón de inactivación del cromosoma X y el carácter y localización de la mutación15. Si el cromosoma X con la mutación en MECP2 es favorablemente inactivado, se observa un fenotipo intermedio. Las variaciones en la región carboxilo terminal de la proteína conducen a un fenotipo menos severo15,23,24. Además, los pacientes con mutaciones de cambio de sentido padecen habitualmente un síndrome menos severo y mejor desarrollo del lenguaje que aquellos con mutaciones sin sentido. Sin embargo, no se reportan niveles de afectación diferentes cuando las mutaciones ocurren en los dominios MDB o TRD, aunque sí se ha comprobado que cuando ocurren en estas regiones conducen a fenotipos mucho más graves ya que disminuyen la estabilidad de la MECP2 in vivo a la vez que afectan la capacidad de dicha proteína de reprimir la transcripción14,25. Bartholdi et al.26 reportaron que las mutaciones en el exón 1 del gen MECP2 producen fenotipos más severos ya que afectan la expresión de la isoforma B la cual es más abundante en el cerebro que la A.
Otro fenotipo asociado a alteraciones en el gen MECP2 es el que se obtiene como consecuencia de la duplicación completa del gen. Este fenómeno afecta fundamentalmente a varones, como se indicó anteriormente, y resulta en una profunda incapacidad intelectual, hipotonía, retraso del desarrollo psicomotor, crisis epilépticas y comportamiento autista, entre otros aspectos15,27,28.
CDKL5LocusEl gen CDKL5 se encuentra mutado en la mayoría de los pacientes afectados con SR con epilepsia precoz y se localiza en el cromosoma X (Xp22.13)3,29 (fig. 2).

El gen contiene 24 exones distribuidos sobre 240kb de ADN genómico30. Los 3 primeros (1, 1a y 1b) no se traducen y constituyen diferentes sitios de inicio de la transcripción29,30 por lo que la región codificante está comprendida entre los exones 2 y 2130. En el año 2011, Fichou et al.31 descubrieron un nuevo exón al cual denominaron 16b por su localización entre los exones 16 y 17.
Acción del genCodifica la proteína cinasa dependiente de ciclina 5 (CDKL5)29.
Producto génicoLa proteína CDKL5 está ampliamente distribuida en todos los tejidos del organismo aunque se encuentra en niveles mucho más elevados en el cerebro, especialmente en el período postnatal y en regiones como el hipocampo y la corteza15,32,33.
Existen 2 isoformas con diferentes regiones no traducidas hacia el extremo 5’ (5’ UTR): la isoforma 1, que contiene el exón 1 y se expresa en gran cantidad de tejidos, y la 2, que contiene a los exones 1a y 1b y se expresa solamente en los testículos y el cerebro fetal. El transcrito principal genera una proteína de 1.030 aminoácidos y 115kDa de peso molecular (CDKL5115). Por otra parte, la CDKL5107, en cuya secuencia codificadora permanece el intrón 18, está ampliamente distribuida en un gran número de especies, incluyendo los ratones, lo cual permite el estudio de las funciones de esta proteína en modelos animales34.
La proteína CDKL5 tiene un dominio catalítico en su extremo amino terminal (aminoácidos 13-297) y es la única, en la familia de las cinasas, que tiene una larga cola (con más de 600 aminoácidos) que no presenta similitud evidente con otros dominios proteicos pero con un alto grado de conservación ya que solo existen pequeñas variaciones en la región más externa de su extremo C-terminal entre las diferentes especies animales. Además de la región de unión al ATP y el sitio activo de la actividad cinasa sobre serina/treonina (aminoácidos 13-43 y 131-143 respectivamente), la proteína se caracteriza por tener un motivo Thr-Xaa-Tyr (TEY) en los aminoácidos 169-171 cuya fosforilación está normalmente involucrada en la activación de la función cinasa de la proteína. También presenta señales de importación y exportación nuclear (NLS y NES respectivamente) en su extremo carboxilo terminal30.
La proteína CDKL5 es una proteína nuclear pero también puede encontrarse en el citoplasma celular. Esta dualidad en cuanto a su localización está estrictamente regulada18,35. Algunos autores proponen que la proteína se ubica inicialmente en el núcleo pero la pérdida de su dominio C-terminal aumenta su expresión a la vez que activa su actividad autofosforiladora, lo cual trae como consecuencia que comience a expresarse en el citoplasma29. Sin embargo, Rusconi et al.36 plantean que la salida de la proteína del núcleo es promovida por una activación del glutamato en el grupo de receptores N-metil-d-aspartato. En el citoplasma, la proteína desempeña funciones tan importantes como la regulación sobre la migración neuronal y la morfología de neuronas y dendritas18,37.
La expresión de la proteína CDKL5 se correlaciona, tanto in vitro como in vivo, con la maduración neuronal. Aun cuando los niveles de la proteína disminuyen en el cerebro adulto, estos son significativamente altos cuando se comparan con los que se encuentran en otros tejidos, por lo que es posible especular que dicha proteína desempeñe un papel fundamental en otras funciones neuronales. Por otra parte, la distribución intracelular de la CDKL5 cambia durante el proceso de maduración neuronal pues se ha comprobado que la fracción nuclear aumenta en el cerebro adulto lo cual sugiere que la misma puede estar involucrada en la plasticidad sináptica. Asimismo, está demostrado que la cascada de señales derivadas de la actividad de esta proteína es fundamental en procesos como el aprendizaje, la ramificación de las dendritas y la formación del citoesqueleto de actina del citoplasma celular30.
Ricciardi et al.32 propusieron que esta cinasa también pudiera estar involucrada indirectamente en el procesamiento del ARN mensajero al fosforilar a las proteínas encargadas del splicing o corte y empalme del ARN mensajero.
Datos clínicos, genéticos y biológicos validan la existencia, tanto in vivo como in vitro, de una estrecha colaboración entre la CDKL5 y la MECP2, aunque aún no pueda definirse correctamente si es la primera la que tiene un control directo sobre la fosforilación y activación de la segunda o, por el contrario, es esta última la que regula la transcripción de la cinasa30,38. No obstante, es indudable la cooperación de ambas proteínas en la regulación de la expresión génica (existe evidencia de que la MECP2 recluta a CDKL5, junto a otras proteínas, para formar un complejo de unión al ADN con actividad cinasa29) lo cual explica la similitud entre los fenotipos derivados de las mutaciones en estos 2 genes. Además, la expresión de la cinasa aumenta considerablemente en la etapa posnatal al igual que ocurre con la MECP2 en el cerebro de ratones adultos18,35.
Tipos de mutaciones y consecuencias de su expresiónEn el gen CDKL5 se han descrito varias mutaciones que incluyen grandes y pequeñas deleciones, mutaciones de cambio de sentido, mutaciones sin sentido, corrimientos del marco de lectura y variaciones que afectan los mecanismos normales de corte y empalme30 (tabla 2).
Algunas mutaciones descritas en el gen CDKL530
| Mutaciones |
|---|
| 39delT; 183delT; 163_166delGAAA; 275_276insAA; 800_801delAT; 838_847del10; 865insA; 903_904dupGA; 964dupA; 1079delT; 1311dupC; 1892_1893dupTA; 2014insC; 2016delC; 2045_2046delAGins18; 2066delC; 2323_2324delGA; 2343delG; 2362-2366delAAGAAA y 2635_2636delCTG20R; A40V; R59X; N71D; I72N; I72T; Q118X; H127R; V132G; L142X; C152F; R158; R175S; R178P; R178W; R178Q; P180L; S196L; E203D;E203X; L220P; L227R; T288I; C291Y; L302F; Q347X; S413X;R550X; R559X; E570X; V718M; Q834X; R952X y R970X |
Fuente: Kilstrup-Nielsen et al.30.
Al analizar la distribución de las mutaciones de cambio de sentido, resulta evidente que se localizan fundamentalmente en el dominio catalítico lo cual confirma la importancia de la actividad cinasa de esta proteína para el correcto desarrollo y/o función del cerebro. Por el contrario, las mutaciones que provocan la formación de proteínas truncadas pueden encontrarse en cualquier lugar del gen y, por tanto, pueden obtenerse proteínas de diferentes pesos moleculares. La relevancia del no bien caracterizado extremo C-terminal de la proteína se sugiere por el hecho de que un gran número de variantes patogénicas involucran esta región30.
Estudios recientes han revelado que solamente una pequeña fracción de las mutaciones descritas en el gen CDKL5 es responsable del fenotipo característico del SR ya que la mayoría son la causa de la encefalopatía epiléptica de comienzo temprano15,39,40. Esto se corresponde con lo reportado por Russo et al.41 quienes identificaron solamente 7 mutaciones en el gen CDKL5 en pacientes con SR con epilepsia precoz: 2 mutaciones de cambio de sentido, 2 en sitios de splicing, un corrimiento del marco de lectura, una mutación sin sentido y una inserción.
Todas las mutaciones asociadas a fenotipos severos causan pérdida de la actividad cinasa de la proteína29.
Correlación genotipo-fenotipoHasta el momento, no se ha podido establecer una clara correspondencia entre genotipo y fenotipo para las diferentes mutaciones descritas en el gen CDKL5. Algunos reportes indican que las mutaciones en el extremo C-terminal originan variantes fenotípicas menos severas que las que ocurren en el dominio catalítico30 y que las mutaciones sin sentido producen síntomas menos graves que las mutaciones de cambio de sentido o en sitios de splicing29. Sin embargo, otros autores apuntan que la mutación no se corresponde con la gran heterogeneidad clínica presente en algunos pacientes. Weaving et al.42, al estudiar a 2 hermanas gemelas idénticas genéticamente con mutaciones en el gen CDKL5, comprobaron que los fenotipos eran significativamente diferentes y sugieren que tales divergencias puedan deberse a la participación de factores ambientales y/o epigenéticos desconocidos que afecten la expresión del gen en estudio30.
A partir de la caracterización de proteínas CDKL5 truncadas en sus extremos C-terminales se ha podido establecer una función reguladora para su larga cola de aminoácidos. De hecho, parece actuar como supresora de la actividad catalítica de la proteína y como moduladora de su localización celular ya que se ha comprobado que los mutantes L879X y R781X confinan a la proteína al núcleo por lo que puede inferirse que la última porción de la proteína tiene como función la localización citoplasmática de la proteína30.
También es importante destacar que, contrario a lo que estaba establecido hasta el momento, reportes recientes indican que no existen diferencias clínicas en cuanto a severidad de los síntomas entre varones y hembras afectados con SR producto de mutaciones en este gen30,43,44.
FOXG1LocusEl gen FOXG1 se encuentra mutado en la mayoría de los pacientes afectados con la variante congénita del SR y se localiza en el cromosoma 14 (14q12)3,45 (fig. 3).

El gen contiene 5 exones y no tiene intrones46. El dominio N-terminal presenta una región rica en prolina y glutamina que es específica de los mamíferos45.
Acción del genCodifica la proteína FOXG1 (antes llamada brain factor 1 o BF-1), que actúa como factor represor de la transcripción3,15,45 al reclutar a las proteínas Groucho y KDM5B que inhiben la expresión de determinados genes15.
Producto génicoLa proteína FOXG1 tiene 476 aminoácidos y 4 isoformas46.
Presenta expresión restringida al cerebro y al tejido testicular5. En el período fetal actúa sobre el neuroepitelio telencefálico, el área nasal de la retina y el nervio óptico. Es fundamental para la correcta regulación del desarrollo del cerebro y del telencéfalo a partir del cual se forma la corteza cerebral y los ganglios basales47. También se expresa en el cerebro maduro y Florian et al.48 sugieren que promueve la supervivencia de neuronas posmitóticas diferenciadas15. En las regiones frontales favorece la proliferación de ciertos precursores e inhibe la neurogénesis prematura8,9.
Tipos de mutaciones y consecuencia de la expresiónSolamente se han descrito 7 mutaciones en el gen FOXG1 causantes de la variante congénita del SR y en todos los pacientes analizados han constituido mutaciones de novo45,46. Todos los individuos afectados se caracterizan por la ausencia de un período normal de desarrollo luego del nacimiento y por una severa microcefalia15,47,49,50.
Kortum et al.51 reportaron que la mayoría de las mutaciones son cambios de un único nucleótido aunque también se encuentran deleciones y duplicaciones (tabla 3).
Mutaciones en el gen FOXG1 causantes de la variante congénita de SR y las consecuencias de su expresión expresión
| Mutación | Consecuencia de la mutación |
|---|---|
| TRP255TER | Afecta a las 4 isoformas de la proteínaOrigina codón de terminación |
| 969delC | Afecta a las 4 isoformas de la proteína |
| TRY208TER | Origina codón de terminación |
| PHE215LEU | – |
| TRP308TER | Origina codón de terminación |
| TRP400TER | Origina codón de terminación. Conduce a fenotipos menos graves ya que se ha comprobado que pacientes con esta mutación tienen actividad residual de la proteína Foxg17 |
| 460_461dupG | Es una duplicación de G después de 7 nucleótidos G ubicados de manera consecutiva. Es la mutación que se encuentra con mayor frecuencia en los pacientes con variante congénita del SR por lo que se piensa que este sitio constituya un punto caliente para mutaciones en gen FOXG126 |
La búsqueda de una relación genotipo-fenotipo en los pacientes con mutaciones en el gen FOXG1 resulta difícil por los pocos casos descritos hasta el momento. La evolución de estos niños no ha permitido relacionar la localización de la mutación en el gen con la gravedad de las manifestaciones clínicas, ya que pacientes con mutaciones condicionantes de proteínas truncadas en los mismos dominios activos, y en localizaciones muy próximas, presentan expresiones clínicas muy diferentes3.
Algunas estrategias terapéuticasHasta el momento no existe un tratamiento directo y curativo para el SR. Numerosas estrategias terapéuticas han sido exploradas en modelos animales y se ha comprobado que, mayoritariamente, la disminución en la severidad de los síntomas depende de cuán temprano se administren los diferentes medicamentos. Fármacos como la despiramina, el factor de crecimiento similar a la insulina tipo 1 y los agonistas parciales de los receptores TrkB son empleados para disminuir los síntomas y prolongar las expectativas de vida de los diferentes tipos de mutantes52.
El entendimiento incompleto de la adecuada correlación entre genotipo y fenotipo para los diferentes genes involucrados y los mecanismos de acción de la proteína MECP2 continúan impidiendo el desarrollo de terapias racionales para el SR, por lo que una estrategia basada en el tratamiento directo sobre las causas subyacentes del mismo resultaría realmente atractiva. En este sentido, la terapia génica, o sea, la inserción de copias de la variante nativa del gen MECP2 en células con ausencia de la MECP2 normal, representa una opción potencial de tratamiento para los pacientes con SR ya que se ha demostrado que, en ratones, la activación posnatal de MECP2 conduce a un fenotipo menos agresivo lo cual indica que aspectos característicos del síndrome pueden ser reversibles y potencialmente prevenibles si son tratados tempranamente53,54. Por otro lado, la inactivación de este gen en ratones adultos conduce a un fenotipo similar al del SR lo cual sugiere la participación de la proteína en el funcionamiento de las neuronas adultas55. Por tanto, para que la terapia génica sea realmente efectiva no solo es necesario garantizar la expresión de copias funcionales de MECP2 en el cerebro sino también mantener los niveles correctos de la proteína derivada exógenamente55.
En estudios recientes, Gadalla et al.56 demostraron que el tratamiento con AAV9/MECP2 (gen MECP2 de origen humano insertado en un vector viral), administrado directamente al cerebro de ratones machos con dicho gen inactivado, resulta en la expresión de una MECP2 normal a niveles que llevan a una reducción significativa de la severidad fenotípica así como a una prolongación sustancial de la expectativa de vida. Los autores concluyen que futuros estudios en ratones hembras heterocigóticas, además de la necesidad de la aparición de herramientas que permitan detectar con mayor sensibilidad las diferentes variantes fenotípicas en esta especie animal, son aspectos fundamentales a desarrollar antes de que pueda considerarse la aplicación de este procedimiento en seres humanos56.
Por su parte, Schaevitz et al.57 realizaron un estudio para determinar la eficacia de la acetil-L-carnitina (ALC) en ratones con desórdenes del neurodesarrollo similar al SR ya que es bien conocido que la misma produce un mejoramiento de las funciones motoras y cognitivas en un gran número de modelos animales a través de mecanismos que incluyen la expresión de neurotrofinas y el mejoramiento de las funciones mitocondriales58. Los autores comprobaron que la ALC es capaz de atenuar gran cantidad de síntomas en los mutantes MECP2 en mayor medida que la mayoría de las terapias farmacológicas utilizadas hasta el momento y sin efectos adversos aparentes. Además, este es el primer estudio en reportar un mejoramiento significativo en la morfología de las dendritas (en el SR, la longitud y complejidad de las mismas están disminuidas en regiones del cerebro como la corteza y el hipocampo) lo cual puede ser consecuencia de varias de las funciones de la ALC tales como la producción de un incremento en la síntesis y liberación de acetil-colina, glutamato y dopamina58. Por otra parte, Schaevitz et al. hacen especial énfasis en la importancia de la administración temprana de ALC (durante el período crítico de desarrollo cortical y de formación de sinapsis) ya que comprobaron que la suplementación con ALC se volvía menos efectiva a medida que progresaban los síntomas. Concluyen que la administración de ALC en el período de desarrollo equivalente al tercer trimestre de embarazo en humanos (antes del nacimiento y en etapas tempranas luego de este) permitiría la obtención de mejores resultados en la prevención del desarrollo de los síntomas clásicos del SR por lo que debería considerarse el diagnóstico prenatal de mutaciones en el gen MECP2 y de los niveles de expresión de la proteína como parte de los programas de detección de enfermedades genéticas57.
Consideraciones finalesEl diagnóstico del SR, tanto en su forma clásica como en sus variantes atípicas, se basa en los criterios clínicos. No obstante, los avances en la biología molecular y en la genética en particular han abierto el abanico de posibilidades diagnósticas a las diferentes formas clínicas que antes quedaban sin clasificar, a la vez que el análisis molecular permite confirmar el criterio clínico y aportar información en cuanto al pronóstico del paciente.
En la revisión realizada se encontraron 1.348 artículos relacionados con este síndrome, de los cuales se seleccionaron 42 dedicados exclusivamente a las bases moleculares del mismo, publicados en el período comprendido entre el 2010 y 2013, representando el 72% de los artículos citados. Otros 16 artículos fueron revisados.
En toda la bibliografía consultada se reportaron 3 genes causantes del síndrome (MECP2, CDKL5, FOXG1) con numerosas mutaciones relacionadas.
Se describió que el gen MECP2 está mutado en el 80% de los pacientes con SR clásico así como en el 40% de los afectados con alguna de sus formas atípicas (regresión tardía y lenguaje conservado mayoritariamente) y se han identificado más de 225 mutaciones en el mismo; de ellas solo 8 se encuentran en el 70% de los pacientes con SR que cumplen completamente los criterios de consenso para el diagnóstico. Por otra parte, el SR con epilepsia precoz y la variante congénita se deben, fundamentalmente, a variaciones en los genes CDKL5 y FOXG1 respectivamente.
En la actualidad no existe un tratamiento directo y curativo para el SR aunque numerosas estrategias terapéuticas han sido exploradas en modelos animales. Muchos son los estudios que intentan el uso de la terapia génica o el aporte de algunas sustancias desde etapas tempranas de la vida, pero a pesar de los prometedores resultados, aún diferentes factores imposibilitan el uso y aplicación de estos procedimientos en humanos.
Una estrecha colaboración entre clínicos, genetistas e investigadores facilitará el diagnóstico de los pacientes con SR, la identificación de las distintas mutaciones y abrirá nuevas puertas en el manejo de esta infrecuente afección.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiamiento, estudios animales y sobre la ausencia de conflictos de intereses según corresponda.
Otros sitios consultados:
- 1.
ORPHANET (Disponible en: http://www.ojrd.com)
- 2.
GeneMap (Disponible en: http://www.omim.org/search/advanced/geneMap)
- 3.
Genetests (Disponible en: www.genetests.org)
- 4.
Proteins (Disponible en: http://www.ncbi.nlm.nih.gov/protein/)
- 5.
Gene (Disponible en: http://www.ncbi.nlm.nih.gov/gene/)
- 6.
Gene Reviews (Disponible en: www.genereviews.org)
- 7.
RettBASE: IRSF MECP2 Variation Database (Disponible en: http://mecp2.chw.edu.au/)
- 8.
OMIM (Disponible en: http://www.ncbi.nlm.nih.gov/omim/)
