


La morfea o esclerodermia localizada juvenil (ELJ) es una enfermedad autoinmune, inflamatoria, crónica, lenta y progresiva del tejido conectivo, de causa desconocida, que afecta preferentemente la piel y los tejidos subyacentes.
ObjetivosComunicar un caso de esclerodermia localizada juvenil en una escolar, y contribuir a un diagnóstico y tratamiento oportuno de esta patología.
Caso clínicoNiña de 8 años con placas induradas hipopigmentadas, de distribución lineal en la extremidad superior derecha de 2 años de evolución y placas induradas hiperpigmentadas de textura acartonada, con áreas de piel adelgazada, blanquecina y edema en la pierna y el tobillo. Los elementos clínicos y los exámenes de apoyo diagnóstico, incluyendo la histología, fueron compatibles con ELJ lineal, panesclerótica. Se inició tratamiento inmunosupresor y simultáneamente realizó fisioterapia y terapia ocupacional intensivas.
ConclusionesPresentamos un caso de ELJ de tipo lineal y panesclerótico, en el que hubo retraso de 2 años en el diagnóstico, no obstante la respuesta al tratamiento inmunosupresor fue favorable según lo esperado.
Morphea or juvenile localised scleroderma (JLS) is an autoimmune, inflammatory, chronic, slowly progressive connective tissue disease of unknown cause that preferably affects skin and underlying tissues.
ObjectiveTo report a case of Juvenil Localised scleroderma in an 8-year old girl, contributing to an early diagnosis and treatment.
Clinical caseThe case is presented of an 8 year-old girl who presented with indurated hypopigmented plaques, of linear distribution in the right upper extremity of two years onset, together with papery texture hyperpigmented indurated plaques with whitish areas of thinned skin in right lower extremity, and leg and ankle swelling. The clinical features and diagnostic tests, including histology were compatible with linear and pansclerotic JLS. She started with immunosuppressive therapy, physiotherapy, and occupational therapy.
ConclusionsWe report a case of linear and pansclerotic ELJ type, in which there was a 2 year delay in diagnosis, however the response to treatment was positive as expected.
Los síndromes de esclerodermia juvenil son enfermedades autoinmunes multisistémicas1, cuya característica común es la presencia de cambios cutáneos escleróticos y comienzo antes de los 16 años de edad. Existen 2 categorías: la esclerodermia juvenil localizada (EJL) o morfea, en que hay preferentemente compromiso cutáneo sin compromiso vascular sistémico o de órganos internos, y la esclerosis sistémica, en la que hay esclerosis cutánea difusa y compromiso de órganos internos2. Aunque la EJL es poco frecuente, en niños es más común que la esclerosis sistémica en una relación de 10:13,4.
En Chile no hay registro de series de pacientes pediátricos reportados. La baja incidencia (uno por millón de niños/año)4 podría explicar el escaso número de pacientes notificados5. Zulian et al. (2006) publicaron las características clínicas y epidemiológicas de 750 niños con ELJ de 270 centros reumatológicos y dermatológicos de todo el mundo, modificando la clasificación de Peterson (Clasificación de Mayo) que incluía 5 formas clínicas (en placas, generalizada, bulosa, lineal y profunda) por: circunscrita, lineal, generalizada, panesclerótica y mixta (Clasificación de Padua), enfatizando el retraso en el diagnóstico como consecuencia del comienzo insidioso y la falta de conocimiento de la enfermedad5,6. El 12,1% de los pacientes tenía antecedentes familiares de enfermedades autoinmunes6–8. La relación mujer/hombre fue de 2,4:19.
De los 5 subtipos descritos en la clasificación de Padua la esclerodermia lineal (EL) es el más común (65%) en pediatría. Se caracteriza por uno o más trayectos lineales que involucran típicamente una extremidad superior o inferior. Le sigue en frecuencia la morfea circunscrita (26%), generalizada (17%), panesclerótica (2%) y mixta (15%) que combina EL con otros subtipos6.
Presentamos una niña cuyo diagnóstico e inicio de tratamiento se realizó 2 años después del comienzo de las lesiones cutáneas, con el objetivo de difundir los conceptos y elementos clínicos que permitan que los médicos y profesionales de la salud puedan sospechar precozmente y derivar a los especialistas dermatólogo y/o reumatólogo oportunamente.
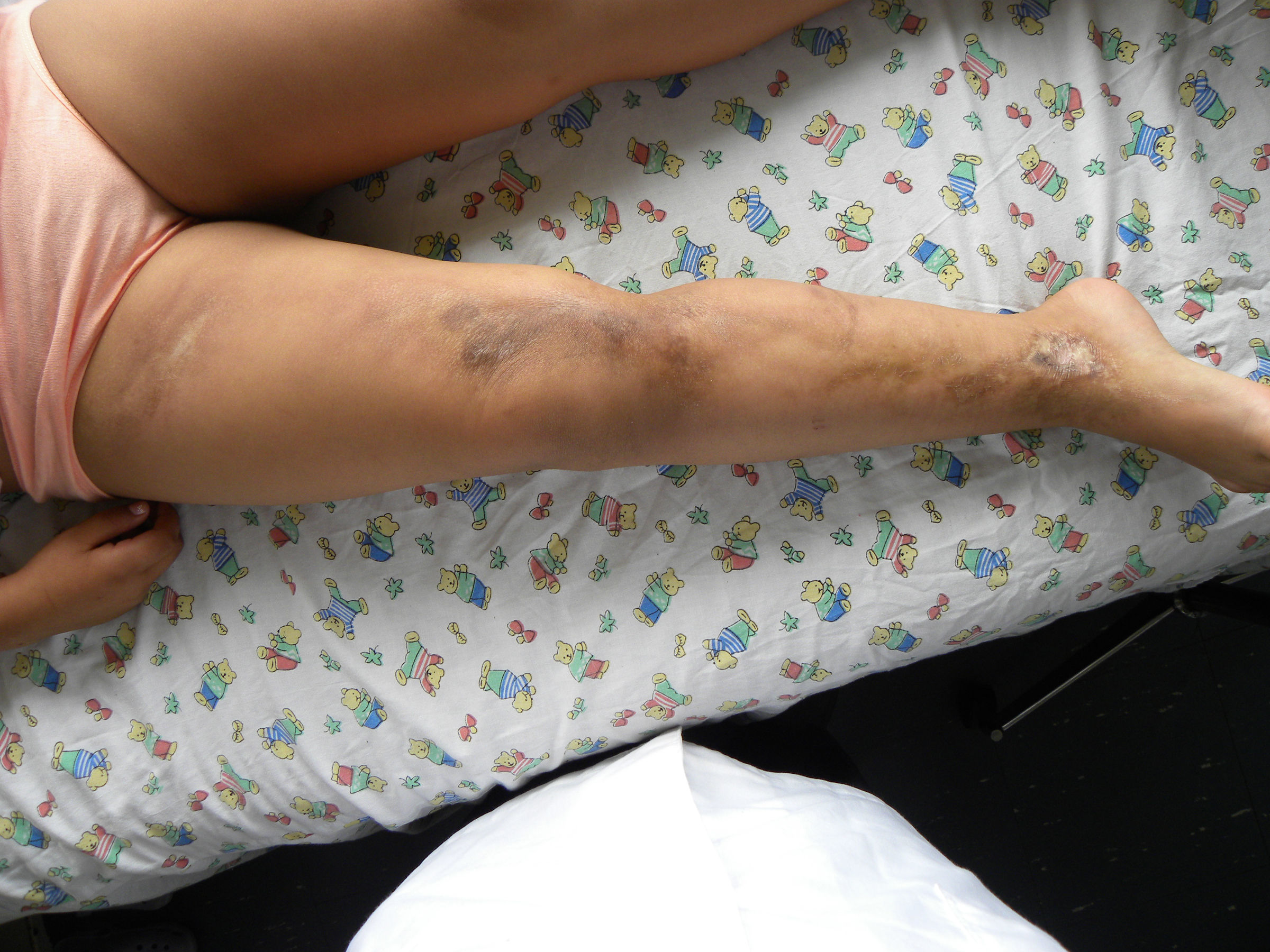
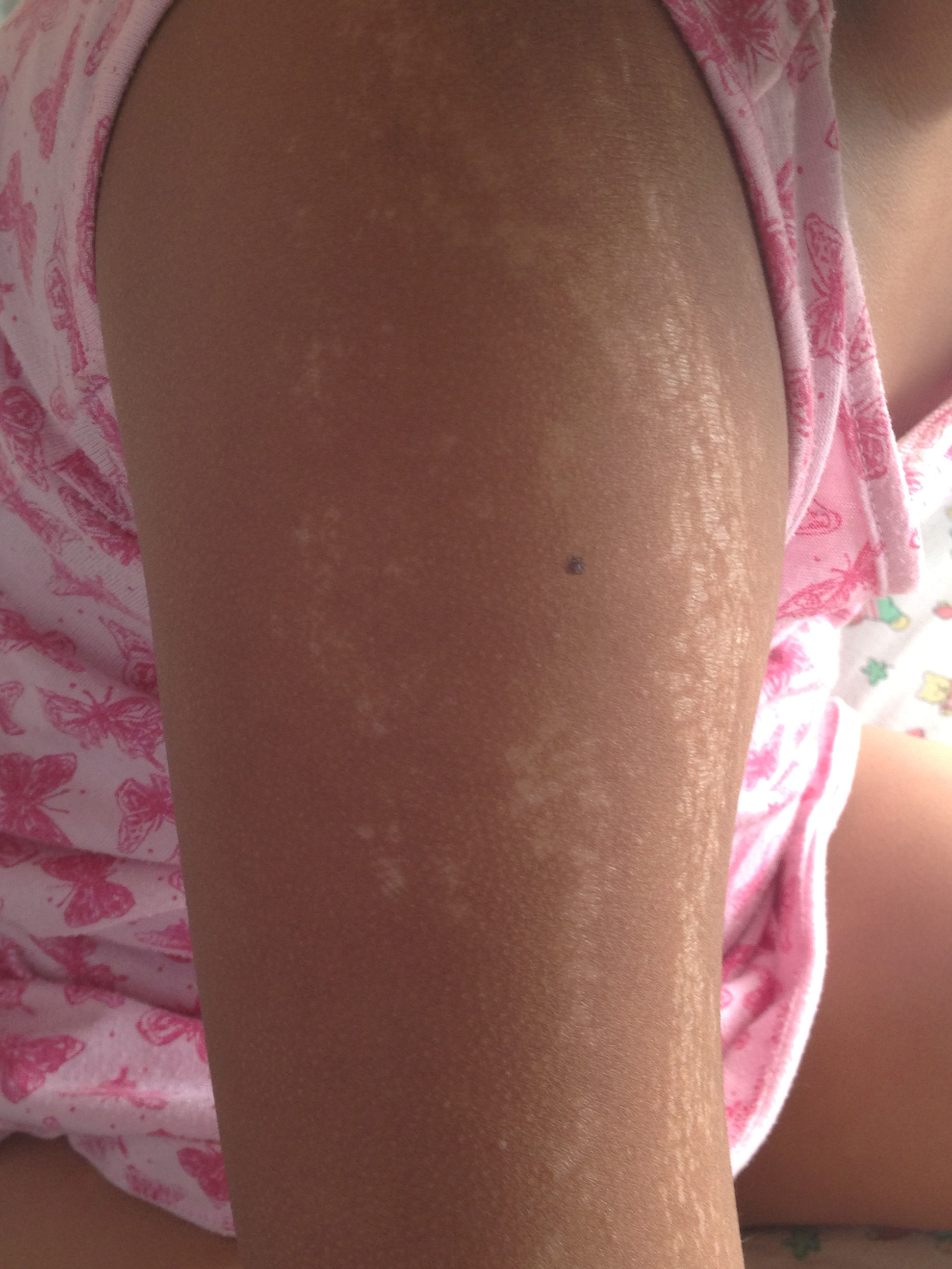
Caso clínicoNiña de 8 años, previamente sana, sin antecedentes familiares de enfermedades crónicas ni autoinmunes. Consultó en atención primaria por lesiones de piel de 6 meses de evolución; inicialmente presentaba 3 máculas hiperpigmentadas de 2 a 3cm de diámetro en la región suprarrotuliana derecha, que progresaron en 4 meses abarcando desde el pliegue inguinal hasta el tobillo, adquiriendo una textura acartonada con áreas deprimidas de piel adelgazada, brillante, de color blanco y edema en el tobillo, sin dolor ni compromiso funcional de la extremidad inferior afectada. Recibió terapia con clotrimazol y betametasona tópicas sin efecto (fig. 1). Posteriormente apareció una mácula eritematosa de 3cm de diámetro en el eje mayor, adyacente al labio mayor derecho. En la extremidad superior derecha presentaba placas induradas hipopigmentadas punteadas, de distribución lineal desde la muñeca al hombro (fig. 2), y varias máculas faciales redondas, simétricas, hipopigmentadas de 2 años de evolución.


Ingresó en el hospital de derivación para evaluación multidisciplinaria. Los exámenes revelaron: hemograma, pruebas tiroideas, inmunoglobulinas séricas, complemento y reactantes de fase aguda normales, radiografía de extremidad inferior derecha, radiografía de tórax, ecocardiografía y ecografía abdominal normales. Estudio inmunológico: ANA 1/40 homogéneo, factor reumatoide positivo 80U, ANCA C negativo, ENA 0,98 (positivo >1,10) y anticoagulante lúpico negativo.
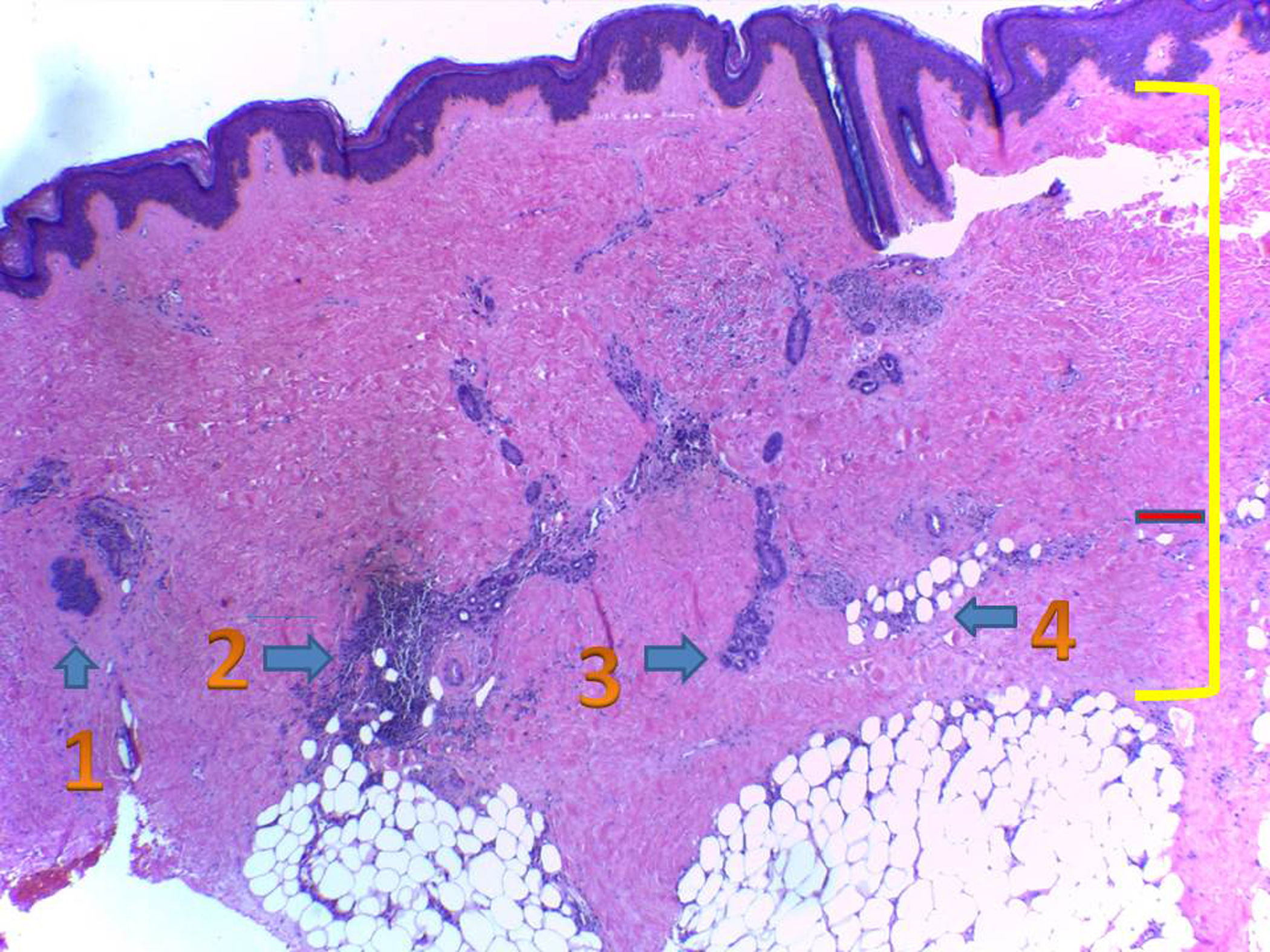
La biopsia cutánea mostró: epidermis de espesor conservado. A nivel subepitelial se evidenció fibrosis de la dermis papilar y reticular, compuesta por fibras colágenas engrosadas, que se extendió hacia el subcutáneo, rodeando las glándulas ecrinas. Escaso tejido adiposo en la periferia y un moderado infiltrado linfohistiocítico, en partes también perivascular, con algunas células plasmáticas. Folículos pilosos atróficos, escasos. Tejido adiposo subcutáneo desplazado hacia la profundidad, con lobulillos separados por septos fibrosos engrosados (fig. 3).

Histopatología de lesión de piel:
1. Atrofia de folículo piloso.
2. Infiltrado inflamatorio linfocitario, con presencia de algunas células plasmáticas alrededor de las glándulas ecrinas, donde aún quedan algunos adipocitos.
3. Glándulas ecrinas con total reemplazo del tejido adiposo por colágeno.
4. Tejido adiposo subcutáneo rodeado por haces de colágeno.
La línea roja marca el espesor dérmico normal y la línea amarilla la expansión del colágeno, con haces engrosados, eosinofílicos.
Se inició tratamiento con prednisona oral 2mg/kg/d por 7 días y en dosis decreciente un mes, asociado a metotrexato oral (14,5mg/m2), con el cual continuó por 16 meses, mejorando la textura de las lesiones de la extremidad inferior, sin cambio significativo en la extremidad superior, sin aparición de nuevas lesiones. Simultáneamente realizó fisioterapia y terapia ocupacional intensivas.
DiscusiónLa EJL o morfea es una enfermedad del tejido conectivo de etiología desconocida, reportada desde 1753. Se caracteriza por depósito excesivo de colágeno, con engrosamiento e induración de piel y tejido subcutáneo, produciéndose cambio de la coloración y consistencia de la piel1–7.
Clínicamente existen 2 fases, una precoz de mayor actividad, durante la cual pueden aparecer lesiones caracterizadas por un borde inflamatorio violáceo o anillo lila, con piel indurada en el centro y en el borde. Durante esta etapa las lesiones pueden expandirse y aparecer nuevas lesiones10,11. Los hallazgos histológicos consisten en infiltrado perivascular, predominantemente de linfocitos, con escasas células plasmáticas y eosinófilos en la dermis reticular profunda y tejido subcutáneo, acompañado de haces engrosados de colágeno, disminución de fibras elásticas y células endoteliales aumentadas de tamaño. En la etapa tardía se aprecia un aumento de grosor de la piel, especialmente en el centro de la lesión, dejando a veces un centro esclerótico hipopigmentado, y la histología revela colágeno engrosado que reemplaza el infiltrado inflamatorio previo, lo que conduce a atrofia de las glándulas ecrinas y folículos pilosos. Además, los capilares tienen una pared fibrótica y una luz estrecha10,11. Estos elementos se correlacionan clínicamente con atrofia dérmica y subdérmica, expresada como adelgazamiento cutáneo, y en los casos más severos depresión o concavidad de la zona afectada, e induración como se observa en las figuras 1 y 3.
La EL es el subtipo más común, caracterizándose por una línea o banda que involucra la dermis, la hipodermis y frecuentemente el músculo subyacente, el tendón y el hueso. Los sitios más comunes son las piernas, los brazos, el hueso frontal y el tronco5,11,12. En las extremidades pueden atravesar una articulación, y si se esclerosan y alteran los músculos y los tendones producir contractura articular y disfunción de la extremidad. Si se afecta la placa de crecimiento epifisiario habrá acortamiento permanente de la extremidad. Se han reportado hasta un 30-50% de complicaciones ortopédicas. Aproximadamente el 25% de los pacientes con EL tiene compromiso de cabeza y cuero cabelludo, denominada «en golpe de sable» debido a la depresión que simula la cicatrización de una herida por ese mecanismo12. En estos casos puede ocurrir compromiso neurológico y ocular de variada severidad13.
La morfea circunscrita o en placa es el subtipo más frecuente en adultos. Se caracteriza por una o más lesiones redondeadas, induradas, de 1-3cm de diámetro, generalmente en el tronco o en la región proximal de las extremidades13,14. La morfea generalizada, definida por más de 3 lesiones en placas, de más de 3cm, que van confluyendo y afectando varias áreas anatómicas, preferentemente en el tronco, es muy infrecuente15.
La morfea panesclerótica afecta al 1-2% de los pacientes y es incapacitante. Inicialmente involucra las extremidades, pero puede migrar a otras localizaciones como el tronco, la cara y las áreas distales de los dedos y las uñas. Ocurre una fibrosis rápidamente progresiva de la dermis, subcutáneo, fascia y músculo con compromiso ocasional de hueso. Este proceso lleva a contracturas articulares, atrofia muscular, ulceraciones cutáneas y, si compromete el tórax, a insuficiencia respiratoria. Puede desarrollarse carcinoma de células escamosas en las áreas de herida crónica6,14,15. La morfea mixta se define por la presencia de más de un subtipo clínico, siendo más frecuente la asociación de EL y en placas6,13,14. Las manifestaciones extracutáneas ocurren hasta en un 25% de los pacientes en el curso de la enfermedad. Es recomendable evaluación oftalmológica, que es mandatoria si la EL afecta la cabeza13,15.
El diagnóstico de EL se basa en el cuadro clínico y, en ocasiones, como en nuestro caso, es apoyado por la biopsia de piel y tejidos subcutáneos. La velocidad de eritrosedimentación puede estar aumentada cuando hay inflamación activa. El factor reumatoideo, positivo en nuestro caso, puede estar presente en el 25-30% de los pacientes, y títulos mayores se han asociado a compromiso cutáneo más severo o afectación articular16. Anticuerpos antinucleares pueden estar presentes en cualquier tipo de morfea, con una frecuencia entre el 23-73%. Anticuerpos anticardiolipinas, más frecuentemente presentes en adultos, solo se encuentran en un 13% de los niños15.
Para evaluar la actividad de la morfea y monitorizar el seguimiento las herramientas disponibles son: iconografías de lesiones, ultrasonido y resonancia magnética, con diferentes ventajas y desventajas, así como sensibilidad y especificidad. Existen también puntuaciones clínicas computarizadas de compromiso de la piel (puntuación cutánea de Rodnan modificada) y la prueba de estudio cutáneo de escleroderma localizado (LoSCAT, por sus siglas en inglés) que combina actividad de la enfermedad y parámetros de daños, los que son de resorte del especialista15,16.
El diagnóstico diferencial es con otras lesiones despigmentadas como vitíligo, liquen escleroso y atrófico, micosis fungoide hipopigmentada, pitiriasis liquenoide crónica, hipopigmentación postinflamatoria, tiña versicolor y pitiriasis alba, así como hiperpigmentadas, postinflamatoria, medicamentosa o metabólica e induradas como paniculitis, síndrome de piel dura, fascitis eosinofílica, dermopatía fibrosante y escleredema.
El tratamiento depende de la severidad de la enfermedad, que considera el subtipo, compromiso potencial de tejidos más profundos, localización de las lesiones y estado de actividad de la enfermedad17.
Las lesiones superficiales y circunscritas pueden desaparecer en 3-5 años sin intervención. Si no hay aumento de las lesiones, eritema u otro signo de actividad, monitorizar la lesión es aceptable. Si hay signos de enfermedad activa una lesión única, superficial, en una localización no cosmética, se puede tratar tópicamente con corticoides, inhibidores de la calcineurina, imiquimod, análogos de la vitamina D o fototerapia con luz ultravioleta17. En las formas moderadas o severas, como la de nuestra paciente, hay consenso en el uso de metotrexato en conjunto con corticoides orales o pulsos de metilprednisolona, tratamientos que han probado ser efectivos, con escasos efectos adversos18–21. La duración del tratamiento con metotrexato no está completamente dilucidada. Se ha usado por períodos prolongados de hasta 2 años sin complicaciones, y el consenso es tratar al menos 2 años, ya que usos en menor tiempo favorecen las recaídas18–21.
Publicaciones recientes han enfatizado las dificultades de acceso, por falta de especialistas, así como los errores de diagnóstico y tratamiento en etapas iniciales de la enfermedad22–24, lo que ocasiona retardo en la referencia y tratamiento específico. Lo óptimo es el manejo multidisciplinario: reumatólogo, dermatólogo, fisiatra, kinesiólogo y terapeuta ocupacional.
ConclusionesPresentamos un caso de EJL de tipo lineal y panesclerótico, que podría corresponder, según la clasificación de Padua, a una forma mixta, confirmado por histopatología, con exámenes de laboratorio compatibles, en el que hubo retraso de 2 años en el diagnóstico. La respuesta al tratamiento inmunosupresor fue favorable según lo esperado.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
