


Los quistes de la vía biliar o quistes de colédoco (QC) son una patología rara en nuestro medio. La etiología es desconocida, siendo la hipótesis más aceptada las anomalías en la unión biliopancreática.
ObjetivoAnalizar los datos clínicos, diagnóstico y tratamiento de una serie de pacientes diagnosticados de QC y realizar una actualización sobre el tema.
MetodoSe revisaron retrospectivamente los diagnósticos de QC en 20 años en un hospital terciario.
Casos clínicosSe identificaron 4 casos, con predominio del sexo femenino. Rango de edad 16 meses a 4 años. Los signos y síntomas fueron ictericia y coluria (100%), vómitos (75%), dolor abdominal y acolia (50%). Ninguno tuvo masa palpable. La ecografía abdominal orientó el diagnóstico que se confirmó con colangio-resonancia magnética (colangio-RM). Se clasificaron como QC tipo I tres de los casos y uno como tipo IVa. El tratamiento fue quirúrgico, ningún paciente presentó complicaciones hasta la fecha.
ConclusionesLos quistes de las vías biliares son de baja prevalencia. El tratamiento de elección es quirúrgico, requiriendo seguimiento estrecho, dado el riesgo de colangiocarcinoma.
Cysts of the bile duct or choledochal cysts are rare diseases in our area. The aetiology is unknown, with the most accepted hypothesis being a pancreatobiliary maljunction anomaly.
ObjectiveTo analyse the clinical data, diagnosis and treatment of a number of patients with choledochal cyst, as well as presenting an update on this condition.
MethodA retrospective descriptive study was performed on paediatric patients diagnosed with choledochal cyst in the last 20 years in a tertiary hospital.
Case reportsA total of 4 choledochal cyst cases in childhood, predominantly female, are pre- sented. The most frequent reason for consultation was vomiting, and presenting with jaundice and choluria in all cases. Patients with choledochal cyst were classified as type I in 3 cases, and one case of type IVa. In all cases surgical treatment was performed; any patient had complications to date.
ConclusionsCysts of the bile ducts have a low prevalence. The treatment of choice is surgical, requiring close monitoring due to the risk of cholangiocarcinoma.
Los quistes de colédoco (QC) son una afección rara consistente en dilataciones de los conductos biliares, tanto intrahepáticos como extrahepáticos. Es una causa frecuente de ictericia, sin embargo, esta entidad es poco prevalente.
Se han descrito diversas teorías sobre su etiopatogenia; la más aceptada es la expuesta por Babbit en 19691. Según esta, un canal común formado por la unión anómala de los conductos pancreáticos y biliares fuera de la ampolla de Vater condicionaría reflujo pancreaticobiliar. Las enzimas pancreáticas activadas causarían inflamación y deterioro de la pared del conducto biliar, lo cual daría lugar a la dilatación biliar.
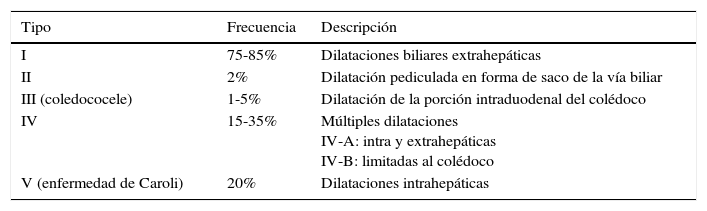
La primera clasificación fue propuesta en 1959 por Alonso-Lej et al.2, dividiendo los QC en 3 grupos, modificada por Todani en 19773, que los clasifica en 5, siendo la más utilizada actualmente (tabla 1).
Clasificación de los quistes de colédoco según Todani (Todani modification of the Alonso-Lej classification)
| Tipo | Frecuencia | Descripción |
|---|---|---|
| I | 75-85% | Dilataciones biliares extrahepáticas |
| II | 2% | Dilatación pediculada en forma de saco de la vía biliar |
| III (coledococele) | 1-5% | Dilatación de la porción intraduodenal del colédoco |
| IV | 15-35% | Múltiples dilataciones IV-A: intra y extrahepáticas IV-B: limitadas al colédoco |
| V (enfermedad de Caroli) | 20% | Dilataciones intrahepáticas |
La tríada clásica (ictericia, masa abdominal palpable y dolor abdominal) pocas veces se presenta completa, siendo el dolor abdominal aislado el síntoma más frecuente.
Se estima una incidencia de 1/13.000 a 1/2.000.000 de recién nacidos vivos4,5, con predominio en poblaciones asiáticas.
El objetivo del presente manuscrito es analizar los datos clínicos, el diagnóstico y el tratamiento de una serie de pacientes diagnosticados de QC y realizar una actualización sobre el tema. Para ello se revisaron retrospectivamente los casos de QC diagnosticados en los últimos 20 años en un servicio de pediatría de un hospital terciario. Se identificaron 4 pacientes, de los cuales se recogieron datos epidemiológicos, clínica, pruebas diagnósticas, tratamiento y evolución.
Casos clínicosCaso 1Paciente de sexo masculino de 16 meses que consultó por cuadro de vómitos no biliosos y febrícula de un mes de evolución, asociado a ictericia cutánea, deposiciones acólicas y coluria en los 3 días previos. En la exploración física presentó coloración ictérica mucocutánea y hepatomegalia de 2cm.
Los exámenes de sangre mostraron un patrón de colestasis, citólisis y aumento de enzimas pancreáticas (tabla 2). La ecografía abdominal reveló hepatomegalia, con colédoco aumentado de calibre en el tercio proximal (9,4mm) y disminución del calibre en tercio distal terminando de forma afilada. Ante la sospecha de enfermedad obstructiva de la vía biliar se inició tratamiento con piperacilina-tazobactam y ácido ursodesoxicólico, junto con dieta exenta de grasa.
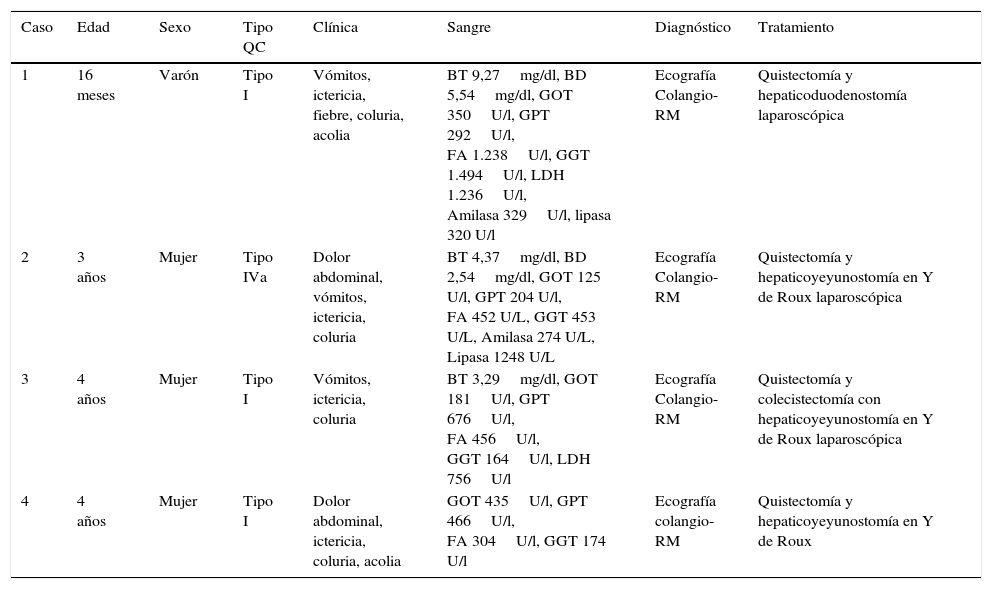
Características clínico-analíticas de los pacientes
| Caso | Edad | Sexo | Tipo QC | Clínica | Sangre | Diagnóstico | Tratamiento |
|---|---|---|---|---|---|---|---|
| 1 | 16 meses | Varón | Tipo I | Vómitos, ictericia, fiebre, coluria, acolia | BT 9,27mg/dl, BD 5,54mg/dl, GOT 350U/l, GPT 292U/l, FA 1.238U/l, GGT 1.494U/l, LDH 1.236U/l, Amilasa 329U/l, lipasa 320 U/l | Ecografía Colangio-RM | Quistectomía y hepaticoduodenostomía laparoscópica |
| 2 | 3 años | Mujer | Tipo IVa | Dolor abdominal, vómitos, ictericia, coluria | BT 4,37mg/dl, BD 2,54mg/dl, GOT 125 U/l, GPT 204 U/l, FA 452 U/L, GGT 453 U/L, Amilasa 274 U/L, Lipasa 1248 U/L | Ecografía Colangio-RM | Quistectomía y hepaticoyeyunostomía en Y de Roux laparoscópica |
| 3 | 4 años | Mujer | Tipo I | Vómitos, ictericia, coluria | BT 3,29mg/dl, GOT 181U/l, GPT 676U/l, FA 456U/l, GGT 164U/l, LDH 756U/l | Ecografía Colangio-RM | Quistectomía y colecistectomía con hepaticoyeyunostomía en Y de Roux laparoscópica |
| 4 | 4 años | Mujer | Tipo I | Dolor abdominal, ictericia, coluria, acolia | GOT 435U/l, GPT 466U/l, FA 304U/l, GGT 174 U/l | Ecografía colangio-RM | Quistectomía y hepaticoyeyunostomía en Y de Roux |
BD: bilirrubina directa; BT: bilirrubina total; colangio-RM: colangio-resonancia magnética; FA: fosfatasa alcalina; GGT: gamma glutamil transpeptidasa; GOT: glutamato oxalacetato transaminasa; GPT: glutamato piruvato transaminasa; LDH: lactato deshidrogenasa.
Permaneció ingresado durante 9 días, con mejoría clínica y de laboratorio. Se realizó colangiorresonancia magnética (colangio-RM) que confirmó el diagnóstico de QC tipo I.
A los 12 meses del diagnóstico, previa mejoría de la clínica y normalización de las enzimas pancreáticas, se realizó cirugía de forma programada con exéresis de QC y hepatoduodenostomía laparoscópica, sin objetivarse complicaciones tras 2 años de la cirugía.
Caso 2Paciente de sexo femenino de 3,5 años que presentó desde el día previo al ingreso vómitos y lumbalgia, junto con orinas colúricas. A la exploración física destacaba ictericia mucocutánea y dolor con defensa en el hipocondrio derecho. Se solicitaron exámenes de sangre, que fueron compatibles con patrón de colestasis, citólisis y elevación de enzimas pancreáticas (tabla 2).
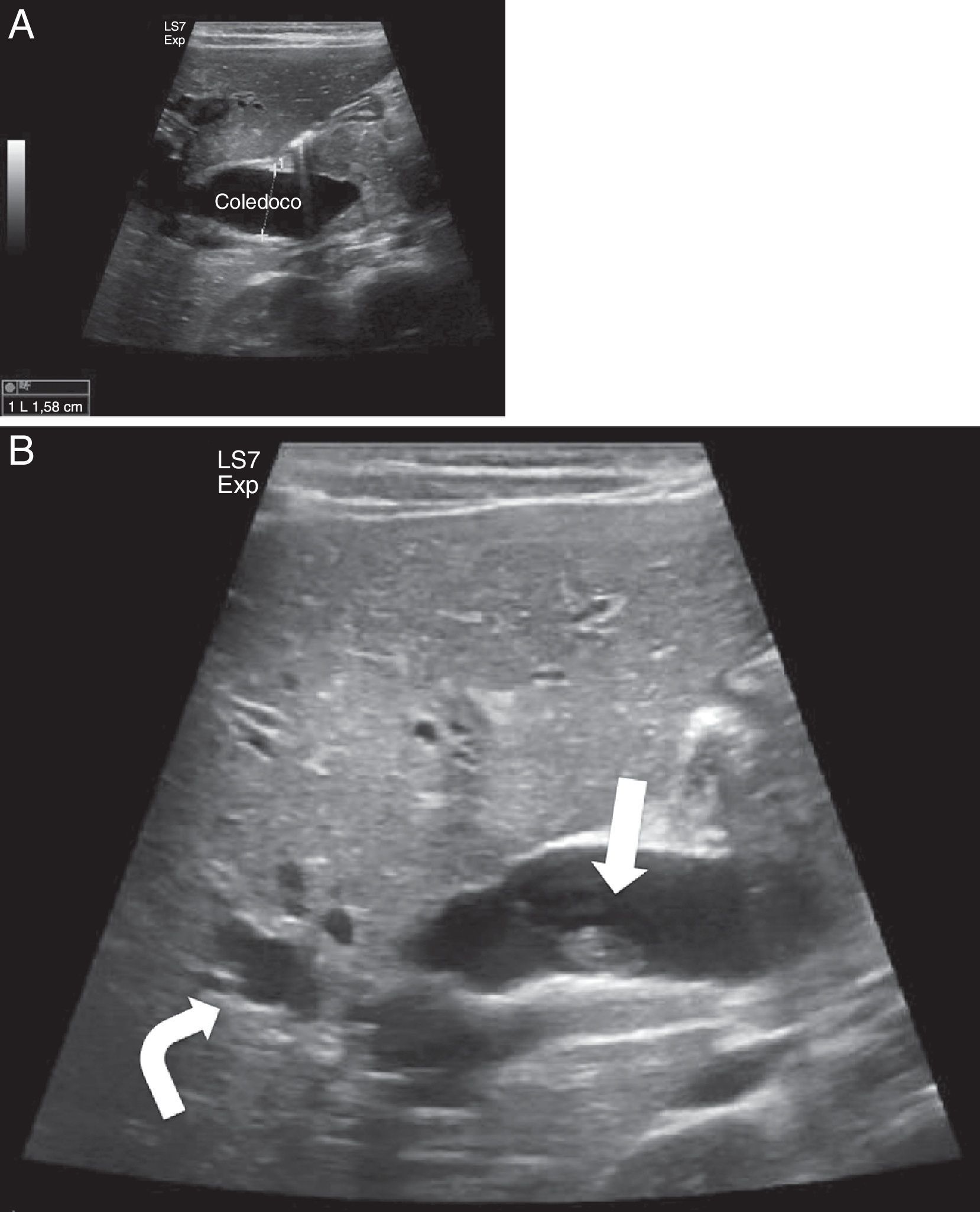
En la ecografía abdominal presentó imágenes compatibles con obstrucción de la vía biliar por presencia de barro biliar y QC tipo IV (fig. 1A y B). Se mantuvo dieta absoluta con fluidoterapia intravenosa hasta lograr mejoría clínica y de laboratorio, y se inició tratamiento con ácido ursodesoxicólico y vitamina K. Se realizó colangio-RM que confirmó QC tipo IVa.
. La vía biliar intrahepática, fundamentalmente la izquierda, muestra un aspecto arrosariado (flecha curva). Ambas imágenes son compatibles con dilatación de la vía biliar extrahepática y probables quistes de vías biliares intrahepáticas (tipo IV).")
Ecografía del caso 2. A. Marcada dilatación de la vía biliar intra y extrahepática, con un colédoco dilatado hasta la papila con un calibre aproximado de 16mm. B. Muestra barro biliar y microlitiasis en la vesícula (flecha). La vía biliar intrahepática, fundamentalmente la izquierda, muestra un aspecto arrosariado (flecha curva). Ambas imágenes son compatibles con dilatación de la vía biliar extrahepática y probables quistes de vías biliares intrahepáticas (tipo IV).
Fue evaluada por un cirujano infantil, realizándose de forma programada al mes del diagnóstico la exéresis del QC y la derivación bilioentérica en Y de Roux por vía laparoscópica. Hasta la actualidad, 20 meses después, no ha presentado complicaciones.
Caso 3Paciente de sexo femenino de 4 años de edad que presentó vómitos de 4 días de evolución y coluria. Al examen físico destacaba ictericia conjuntival y hepatomegalia de 3cm. Se solicitaron exámenes de sangre, destacando patrón de colestasis e hipertransaminasemia (tabla 2). En la ecografía abdominal se objetivó distensión de la vía biliar intra y extrahepática por obstrucción en la desembocadura del colédoco.
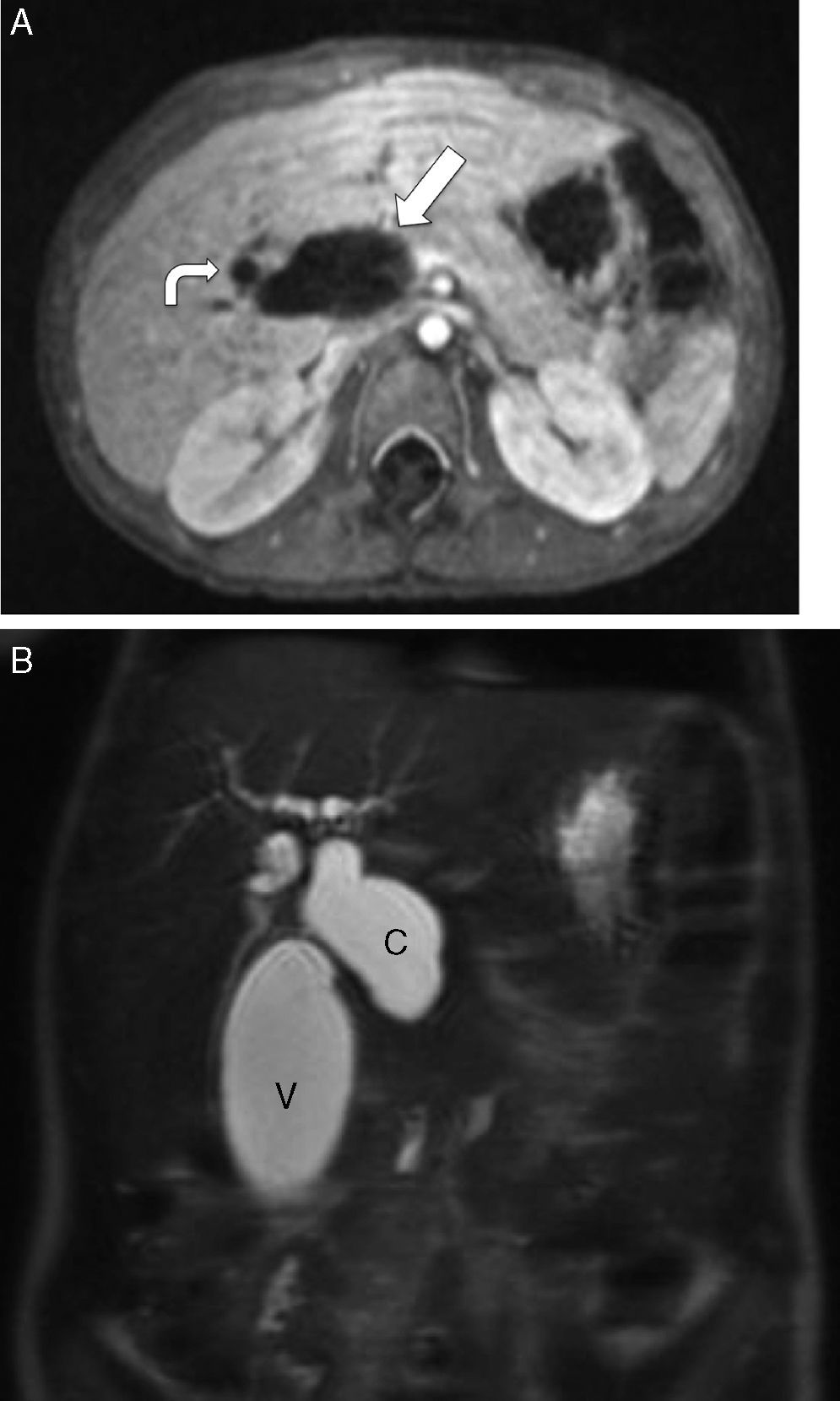
En la colangio-RM se observó dilatación de la vía biliar intra y extrahepática, y de la vesícula, con un colédoco de aspecto sacular con zona filiforme con aspecto de «cola de ratón», hasta alcanzar la zona ampular, con diámetro máximo de colédoco de 2,8cm (fig. 2A y B).
 y escasa dilatación de la vía biliar intrahepática (flecha curva), objetivándose en B un colédoco de aspecto sacular (C) con diámetro máximo de 2,8cm. Dilatación de la vesícula biliar (V). Los hallazgos descritos plantean como primera posibilidad diagnóstica la de un quiste de colédoco tipo 1.")
Colangiorresonancia magnética del caso 3. A. Muestra marcada dilatación de la vía biliar extrahepática (flecha) y escasa dilatación de la vía biliar intrahepática (flecha curva), objetivándose en B un colédoco de aspecto sacular (C) con diámetro máximo de 2,8cm. Dilatación de la vesícula biliar (V). Los hallazgos descritos plantean como primera posibilidad diagnóstica la de un quiste de colédoco tipo 1.
Con los hallazgos descritos unidos a la clínica se planteó como primera posibilidad diagnóstica QC tipo 1. Se realizó de forma programada, a los 2 meses del diagnóstico, quistectomía y colecistectomía con hepático-yeyunostomía en Y de Roux laparoscópica, con adecuada evolución clínica. En controles posteriores con pruebas de imagen 4 años después no se apreciaban cambios respecto a los hallazgos posquirúrgicos.
Caso 4Paciente de sexo femenino de 4 años que presentó dolor abdominal y coluria de 3 días de evolución, asociado a fiebre, anorexia y deposiciones acólicas. En el examen físico destacaba ictericia cutánea. Se realizaron exámenes de sangre que mostraron hipertransaminasemia (tabla 2). En la ecografía abdominal se observaba una imagen quística de 2,5cm compatible con QC tipo I, que se confirmó en la colangio-RM. Presentó mejoría progresiva de los exámenes de sangre tras iniciar una dieta exenta de grasa y tratamiento con ácido ursodesoxicólico.
La paciente presentó colangitis de repetición que motivó varios ingresos hospitalarios para su tratamiento, y obligó a demorar la intervención hasta 2 años después del diagnóstico. La cirugía consistió en quistectomía y colecistectomía con hepático-yeyunostomía en Y de Roux laparoscópica. En una ecografía de control a los 5 años de la intervención presentaba una imagen pseudonodular en interior de asa en Y de Roux, relacionada con litiasis calcificada, sin repercusión sobre la vía biliar.
DiscusiónLos QC son una afección infrecuente de los conductos biliares que se debe sospechar ante la presencia de dolor abdominal, ictericia y masa abdominal palpable. Dos de los 3 hallazgos clínicos aparecen con más frecuencia en población pediátrica (hasta en 85% de los casos), mientras que solo se presentan en el 0-25% de los adultos5,6. En los casos descritos en lactantes el hallazgo más habitual es la presencia de una masa abdominal en el cuadrante superior derecho junto a ictericia; en mayores de un año es más habitual encontrar dolor abdominal, fiebre y vómitos. En 2 series de casos recientes7,8 el dolor abdominal fue el síntoma más frecuente (88-94%), seguido de ictericia (36-58%), vómitos (48%), fiebre recurrente (44%), pérdida de peso (36%) y masa abdominal (8%). En nuestra serie, los signos y síntomas más frecuentes fueron ictericia y coluria (en los 4 pacientes), seguido de vómitos (en 3 de los 4 casos), dolor abdominal y acolia (en 2 de los casos presentados). En ninguno se identificó masa palpable. Durante el tiempo revisado se diagnosticaron 4 casos de QC, 2 de ellos en el último año. El rango de edad fue de 16 meses a 4 años, siendo el único varón de origen asiático.
Ante la sospecha de QC debemos solicitar exámenes de sangre (bioquímica con función hepática, pancreática y renal) y estudio de imagen (ecografía abdominal). En los exámenes aparece alteración de enzimas hepáticas, incremento de amilasa y lipasa junto a leucocitosis. La alteración de las pruebas de coagulación y de función renal indica mayor gravedad9. En los exámenes de sangre realizados a nuestros pacientes, todos presentaron patrón de colestasis y citólisis, y 2 de ellos, además, elevación de las enzimas pancreáticas.
Las pruebas de imagen confirman el diagnóstico. El estudio ecográfico es la primera prueba de imagen a solicitar ante la sospecha de QC10; también es la prueba de elección para vigilar complicaciones postoperatorias. El avance de la ecografía fetal ha llevado al incremento del diagnóstico prenatal, siendo el hallazgo una imagen quística simple al nivel del hemiabdomen superior derecho, generalmente en el segundo o tercer trimestre de gestación.
Tradicionalmente se ha considerado la colangiopancreatografía retrógrada endoscópica (CPRE) la técnica de elección para el estudio preoperatorio; sin embargo, con el progreso de las técnicas de imagen, se ha desarrollado la colangio-RM. Numerosas publicaciones han demostrado eficacia similar en la visualización de la anatomía de las malformaciones de la vía biliar y de la unión pancreaticobiliar entre estas 2 técnicas. Actualmente la colangio-RM puede sustituir a la CPRE, especialmente en niños, en el diagnóstico de esta enfermedad, pues se trata de una exploración no invasiva y con menos complicaciones asociadas, aunque presenta la desventaja de no ser terapéutica. La tomografía computarizada (TC) es útil para mostrar la continuidad del quiste con el árbol biliar, su relación con las estructuras adyacentes y la presencia de malignidad asociada; es mejor que la ecografía en la visualización de la vía biliar intrahepática, la vía biliar distal y la cabeza del páncreas, pero presenta el inconveniente de la radiación. En nuestra serie la ecografía abdominal orientó el diagnóstico en todos los casos, siendo confirmado posteriormente con colangio-RM. No se realizaron otras pruebas de imagen.
El tratamiento depende del tipo de QC. Hay unanimidad en recomendar la cirugía con resección de los QC tipo I8,10. La técnica quirúrgica más utilizada es la quistectomía con reconstrucción de la continuidad de la vía biliar mediante una hepaticoyeyunostomía en Y de Roux9. Frente a la reconstrucción por hepaticoyeyunostomía, en la última década ha ganado popularidad la reconstrucción por hepaticoduodenostomía. Varios estudios demuestran que es una técnica sencilla y segura, con menor tiempo quirúrgico y estancia hospitalaria, sin diferencias significativas en complicaciones posquirúrgicas, excepto una tasa superior de reflujo gástrico11. Sin embargo, aún es pronto para establecer conclusiones debido al corto tiempo de seguimiento y al escaso número de pacientes analizados.
La escisión del quiste por vía laparoscópica ofrece grandes ventajas frente a la cirugía abierta en el tratamiento de los QC12–14. En los quistes tipo III la posibilidad de degeneración maligna es mucho más rara, y en estos casos el tratamiento de elección es la esfinterotomía endoscópica mediante CPRE15. Los QC tipo IVa han sido objeto de controversia con respecto a su tratamiento; la mayor parte de los trabajos publicados recomiendan resección de los quistes extrahepáticos seguida de hepaticoyeyunostomía en Y de Roux9. En cuanto a la enfermedad de Caroli con afectación unilobular, el tratamiento más efectivo es la hepatectomía parcial del lóbulo afectado. En caso de afectación quística difusa, inicialmente el tratamiento debe ser médico con ácido ursodesoxicólico y sales biliares quelantes, pero cuando se producen episodios de colangitis de repetición y cirrosis biliar secundaria descompensada es necesario trasplantar el hígado dañado.
Todos los pacientes revisados en nuestro hospital fueron remitidos a un centro de referencia para evaluación por cirugía pediátrica. En 3 casos el tratamiento fue quirúrgico mediante quistectomía y hepaticoyeyunostomía en Y de Roux, y en el caso restante, el más reciente, se realizó quistectomía y hepaticoduodenostomía, técnica que está ganando popularidad frente a la anterior debido a las ventajas citadas. El tratamiento quirúrgico de QC tiene éxito en más del 90% de los casos, asociándose morbilidad postoperatoria y mortalidad bajas (2,5-27% y 0-6%, respectivamente).
En cuanto al pronóstico, la complicación más temida es el desarrollo de colangiocarcinoma, con riesgo aumentado del 20-30% con respecto a la población general16, por lo que se requiere seguimiento periódico estrecho con el objetivo principal de detectar de forma precoz la posible aparición de este tumor7,10, ya que se han descrito casos de colangiocarcinoma años después de la resección de los quistes17. Los tipos de QC con más predisposición a padecerlo son los tipo I y IV, aumentando el riesgo con la edad18, siendo la edad media de diagnóstico 32 años16,19. Otras complicaciones descritas son: litiasis biliar, litiasis hepática y colangitis (debidas al cúmulo de bilis en las vías biliares), pancreatitis aguda y crónica10,20,21, rotura espontánea22, cirrosis biliar y sus posibles complicaciones debidas a hipertensión portal (sangrado digestivo alto, esplenomegalia y pancitopenia)23,24. Se han descrito otras complicaciones más atípicas como el hallazgo de un quiste hidatídico en el interior del quiste biliar25.
En nuestra serie de casos ninguno ha presentado complicaciones hasta el momento actual, aunque se debe realizar un seguimiento estrecho incluido en edad adulta.
En conclusión, a pesar de su baja prevalencia, ante clínica de vómitos, dolor abdominal e ictericia debemos tener en cuenta el diagnóstico de QC, dado que la tríada clásica como forma de presentación es infrecuente, tal y como se muestra en nuestros pacientes. El diagnóstico definitivo se realiza mediante colangio-RM, aunque la ecografía inicial es orientativa. El tratamiento de elección es quirúrgico, requiriendo un seguimiento periódico estrecho por el riesgo de colangiocarcinoma.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
