




El síndrome de monosomía 1p36 forma parte del grupo de enfermedades conocidas como «enfermedades de baja prevalencia» o «enfermedades raras». El objetivo del presente trabajo es revisar los hallazgos de los principales estudios realizados en niños diagnosticados con el síndrome de monosomía 1p36.
El fenotipo del síndrome de deleción (monosomía) 1p36 delineado desde 1997 incluye rasgos craneofaciales dismórficos: fontanela anterior grande, cejas rectas, ojos hundidos, epicanto, raíz/puente nasal anchos, hipoplasia del tercio medio facial, orejas implantadas anormalmente, filtrum largo y barbilla puntiaguda; alteraciones neurológicas: convulsiones e hidrocefalia (en casos aislados); malformaciones cerebrales observadas en imágenes por resonancia magnética (IRM): ensanchamiento ventricular, ensanchamiento de espacios subaracnoideos, alteraciones morfológicas del cuerpo calloso, entre otras. La IRM evidencia en algunos pacientes atrofia cortical, retraso en la mielinización, áreas multifocales hiperintensas, leucomalacia periventricular y heterotopia periventricular. Estos pacientes cursan con discapacidad intelectual, retrasos en el desarrollo motor, de la comunicación, del lenguaje, en el área personal-social y en la conducta adaptativa. También se observan alteraciones en el sistema auditivo, visual, cardiaco, endocrino, genitourinario, dermatológico y esquelético.
ConclusionesExisten datos de aproximadamente 100 casos en el mundo desde 1981. Esta enfermedad rara es el síndrome más común de microdeleción subtelomérica. La técnica de hibridación in situ con fluorescencia y la técnica de hibridación genómica comparativa (array-CGH) son las que mejor permiten su diagnóstico. Por el momento no existe ningún tratamiento médico efectivo para esta enfermedad.
The Monosomy 1p36 deletion syndrome is part of the group of diseases known as Rare Diseases. The objective of the present work is to review the characteristics of Monosomy 1p36 deletion syndrome.
The monosomy 1p36 deletion syndrome phenotype includes: dysmorphic craniofacial features; large anterior fontanelle, unibrow, deep-set eyes, epicanthus, wide nasal root/bridge, mandible hypoplasia, abnormal location of the pinna, philtrum and pointed chin; neurological alterations: seizures and hydrocephalus (in some cases). Cerebral malformations: ventricular hypertrophy, increased subarachnoid space, morphological alterations of corpus callosum, cortical atrophy, delays in myelinisation, periventricular leukomalacia and periventricular heterotopia. These alterations produce intellectual disability and delays in motor growth, communication skills, language, social and adaptive behaviour. It is Hearing and vision impairments are also observed in subjects with this syndrome, as well as alterations of cardiac, endocrine and urinary systems and alterations at skin and skeletal level.
ConclusionsApproximately 100 cases have been documented since 1981. This rare disease is the most common subtelomeric-micro-deletion syndrome. In situ hybridization with fluorescence (FISH) and array-comparative genomic hybridization (CGH-array) are at present the two best diagnostic techniques. There is currently no effective medical treatment for this disease.
El síndrome de monosomía 1p36 forma parte del grupo de enfermedades conocidas como «enfermedades de baja prevalencia» o «enfermedades raras» (ER). Las ER han sido definidas en el marco de la Comunidad Europea como aquellas con riesgo vital o de carácter crónico debilitante, cuya prevalencia es inferior a 5 casos por 10.000 habitantes. La tasa de prevalencia por debajo de la cual se acepta la calificación de ER varía según el país que otorgue dicha calificación, y oscila entre uno (Australia) y 7 (Estados Unidos) casos por cada 10.000 individuos1.
La mayoría de ER son genéticas, pero no todas tienen esta etiología, también existen ER que tienen un origen infeccioso o parasitario, o incluso pueden ser secundarias a la acción de productos químicos1. La monosomía 1p36 tiene origen genético. Al igual que ocurre con el resto de las ER, algunos casos se diagnostican tardíamente. Los pacientes muestran disfunciones en órganos diversos y enfermedades asociadas, y no cuentan con tratamientos globales, por lo que necesitan atención particularizada de diferentes especialidades médicas.
El síndrome de monosomía 1p36 es una ER poco conocida, existen estudios en los que se presentan algunos aspectos particulares de la enfermedad a nivel de diferentes especialidades médicas. El presente trabajo incluye características del desarrollo evolutivo, así como características clínicas neurológicas, de rasgos craneofaciales dismórficos, cardiacas, esqueléticas, genitourinarias, endocrinológicas, dermatológicas y los genes responsables implicados. También se mencionarán los diagnósticos diferenciales relacionados con el síndrome y aspectos generales del tratamiento.
Para la escritura del presente manuscrito han sido revisadas las bases de datos PubMed, SCOPUS, Google Scholar e ISI web of Knowledge, utilizando las palabras clave: enfermedades raras, monosomía 1p36 y deleción 1p36. Así mismo se ha utilizado el Internacional System for Human Cytogenetics Nomenclature publicado en Cytogenetics and genome Research (2009, 2013) y la base de genes Online Mendelian Inheritance in Man.
Definición y diagnóstico del síndrome monosomía 1p36Es considerado el síndrome más común de microdeleción subtelomérica, no visible mediante cariotipo convencional. Se estima que su incidencia está entre 1:5.000 a 1:10.000 nacimientos2,3, y se han citado cerca de 100 casos en el mundo4. Se han descrito 2 casos en Latinoamérica y 2 en España5-7. El síndrome es causado por la deleción de la región p36 por uno o varios mecanismos genéticos. Se caracteriza por una marcada variabilidad en el tamaño de las deleciones sin puntos de rotura comunes. Se estima que el 52-67% de individuos con este síndrome tienen una deleción terminal en la banda 6 de la región 3 de novo, que el 10-29% tienen una deleción intersticial de tamaño variado, el 7-12% reordenamientos complejos del cromosoma que pueden incluir más de una deleción 1p36 o una deleción 1p36 con una duplicación 1p36, y aproximadamente el 7-12% tienen un cromosoma derivado 18.
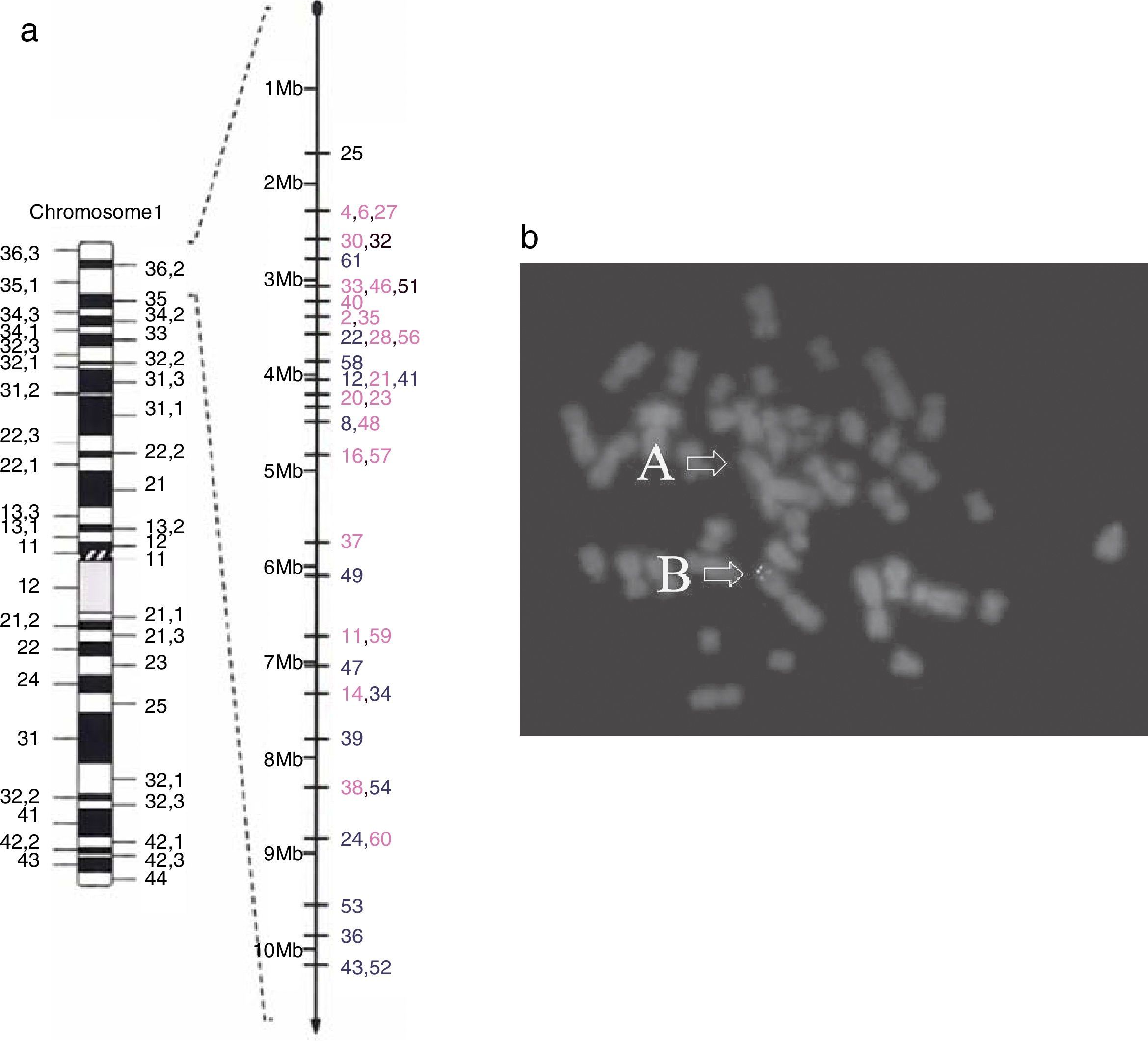
El diagnóstico requiere de análisis genético. Las citogenéticas convencionales no pueden detectar estos diferentes reordenamientos, particularmente los que son cromosomas derivados. La mayoría de las deleciones visibles involucran las bandas teloméricas de los cromosomas. Los reordenamientos de estas regiones son frecuentemente difíciles de identificar por técnicas rutinarias de bandeo. La mayoría de los extremos de los cromosomas se tiñen con GTG-banding (G-banding). Esto es especialmente cierto para la banda terminal del brazo corto del cromosoma 1. Las técnicas que mejor permiten el diagnóstico del síndrome son la técnica de hibridación in situ con fluorescencia (FISH) y la técnica de hibridación genómica comparativa (array-CGH). La técnica FISH, utilizando sondas para las regiones subteloméricas de todos los extremos terminales del cromosoma, excluyendo los brazos cortos acrocéntricos, ha sido de mucha ayuda en la identificación de deleciones terminales que antes eran difíciles de visualizar, incluida la monosomía 1p36 (fig. 1). Algunos estudios refieren el uso de técnicas moleculares en el proceso de diagnóstico, específicamente Multiple Ligation-dependent Probe Amplification (MLPA)6.
, banda 6, región 3 (1p36). B. Estudio de hibridación in situ con fluorescencia (FISH) en fase metafase de un caso estudiado en México. Se puede observar cómo las señales telomérica y subtelomérica están ausentes en el cromosoma con la microdeleción terminal 1p36 (flecha A), y presentes en el homólogo normal (flecha B). Fuente: reproducido de Heilstedt et al.9 (A) y Villarroel et al.5 (B) con autorización.")
Cromosoma 1 y deleción 1p36 de tipo terminal. A. Se presenta la localización de una deleción de tipo terminal en el cromosoma 1, brazo corto (p), banda 6, región 3 (1p36). B. Estudio de hibridación in situ con fluorescencia (FISH) en fase metafase de un caso estudiado en México. Se puede observar cómo las señales telomérica y subtelomérica están ausentes en el cromosoma con la microdeleción terminal 1p36 (flecha A), y presentes en el homólogo normal (flecha B).
Fuente: reproducido de Heilstedt et al.9 (A) y Villarroel et al.5 (B) con autorización.
La técnica array-CGH es también capaz de detectar la complejidad de algunas deleciones propias del síndrome8,9. Un número creciente de pequeñas deleciones intersticiales están siendo identificadas a lo largo de los 30 Mb de ADN que comprenden el cromosoma 1p36 usando esta técnica, lo cual ha permitido determinar que los fenotipos de pacientes con deleciones intersticiales son distintos de aquellos con deleciones terminales, ya que son causados por haploinsuficiencia de un conjunto discreto de genes10,11.
En una paciente española el diagnóstico genético se realizó mediante la técnica MLPA. Tras el análisis de todas las regiones subteloméricas se observó una hemidosis para la sonda que hibrida con la región subtelomérica del brazo corto del cromosoma 1 localizada en el gen tnfrf54 (1p36.33). Asimismo, se validaron estos resultados mediante el análisis por MLPA de otros síndromes microdelecionales, observándose una hemidosis para las 3 sondas que hibridan con la región 1p36, concretamente en los genes TNFRFS4, GNB1 y GABRD, lo que es compatible con una deleción de dicha región en uno de los cromosomas homólogos. Dicho resultado sugirió la presencia de síndrome de deleción 1p36. El método utilizado no excluye la presencia de mosaicismos ni permite detectar otros reordenamientos cromosómicos estructurales (resultados no publicados de Bello y Rodríguez-Moreno).
En cuanto al diagnóstico prenatal y asesoramiento genético en familias que hayan tenido un niño con el síndrome, o en las que se sabe que uno de los padres puede ser portador de un cromosoma reordenado, se pueden combinar técnicas citogenéticas (G-banding, FISH) con muestreo de vellosidades coriónicas entre las 10 y 12 semanas de gestación o con amniocentesis entre las 15 y 18 semanas. El diagnóstico genético preimplantacional se sugiere para parejas que presentan un cromosoma reordenado heredado que pudiera ser un factor de riesgo en relación con el síndrome8.
Primeras aproximaciones al síndrome de monosomía 1p36El primer caso fue descrito en 198112. Se trataba de una niña de 4 años de edad que presentaba retraso mental severo y anomalías congénitas (fontanelas anchas, hipotonía generalizada y murmullo sistólico de grado iii/iv). A partir del análisis por bandeo se demostró su cariotipo balanceado 45,XX,ter rea (1;21) (p36;p13), se sugirió que una deleción submicroscópica del brazo corto del cromosoma 1 podría dar cuenta de las características clínicas de la paciente.
Keppler-Noreuil et al.13 realizaron un estudio en 1995 con 13 pacientes que presentaban pequeñas deleciones terminales que involucraban al cromosoma 1 (1p36.22). Distinguieron 2 tipos diferentes de fenotipias; por un lado, falta de crecimiento y por otro macrosomía. Los autores especularon que estas diferencias podrían ser consecuencia del origen parental o de diferencias en el tamaño de la deleción. Estos pacientes no presentaban deleciones 1p36 puras, la mayoría tenía un doble desbalance segmentario debido a translocaciones desbalanceadas, por lo que sus características clínicas resultantes de la deleción de 1p36 no podían distinguirse de las causadas por el desbalance del otro cromosoma.
El fenotipo del síndrome fue delineado a partir de un estudio realizado con 13 pacientes3. El examen clínico de estos pacientes, mostró que las características más comunes del mismo son: retraso mental, fontanela anterior grande, retraso motor/hipotonía, problemas visuales y auditivos, convulsiones y retraso en el crecimiento. También presentaban características faciales como puente nasal plano, orejas de implantación baja con hélices engrosadas y ojos hundidos.
Durante este mismo año otro estudio describió el caso de una niña de 4 años con mosaicismo por deleción 1p36.33 que presentaba dentro de sus características clínicas: convulsiones (a las 3 semanas del nacimiento), retraso severo del desarrollo, muy pobre adquisición del lenguaje, estrabismo, pérdida auditiva e hipotonía. También presentaba estallidos o arrebatos de mal humor, impulsividad y conductas autolesivas (se mordía las manos cuando estaba enojada) e hiperfagia que incrementaba con la edad (su peso aumentó considerablemente). Tenía frente angosta, ojos hundidos con fisuras palpebrales en forma de «almendra», hipoplasia media facial, boca pequeña girada hacia abajo y un gran paladar ojival. Las orejas pequeñas, bien formadas y en posición normal. Las manos pequeñas, principalmente porque los dedos eran cortos y el quinto dedo con clinodactilia. Los pies pequeños y con dedos cortos. Su evaluación inicial, historia y hallazgos físicos sugerían un posible diagnóstico del síndrome Prader-Willi, pero los análisis citogenéticos lo descartaron14.
Características clínicas de los afectados con el síndrome monosomía 1p36Desarrollo evolutivo a nivel cognitivo, motor, del lenguaje, personal social y conducta adaptativaMuchos autores refieren la presencia de discapacidad intelectual en pacientes con este síndrome; la discapacidad varía desde moderada a profunda y solo se han publicado 3 casos de niños con discapacidad intelectual leve3,4,8,9,15,16. Este síndrome implica alteraciones en el desarrollo motor. Los hallazgos conocidos en esta área se presentan en la tabla 1.
Características del desarrollo motor en niños con síndrome 1p36
| Desarrollo motor | Autores |
|---|---|
| Control cefálico | |
| Para algunos autores se da entre los 5 y los 15 meses de edad en niños con menor retraso, y otros niños en cambio mejoran este control hasta los 2 o 3 años | Battaglia et al.4 |
| Giros | |
| La edad para iniciar giros de supino a prono al parecer se encuentra entre un rango de 6 a 24 meses | Battaglia et al.4 |
| Sedestación | |
| La edad en la que se sientan solos podría estar entre un rango de 7 meses a 3 años, según datos de algunos autores, y entre 9 meses y 5 años y medio para otros. Existe un pequeño porcentaje de niños que pasados estos rangos de edad continúan requiriendo de asientos con apoyo especial, ya que no logran sentarse sin ayuda | Battaglia et al.4 |
| Marcha autónoma | |
| Empiezan a caminar de manera independiente dentro de un rango de edad de 2 a 7 años | Battaglia et al.4 |
| Coordinación | |
| Se observan dificultades de coordinación a nivel de la marcha y de coordinación muscular al tragar | Heilstedt et al.9 |
| Reish et al.17 | |
| Battaglia et al.18 | |
| Tono | |
| El 85% de sujetos presenta hipotonía muscular, más marcada durante los primeros años de vida | Heilstedt et al.9 |
| Battaglia et al.18 | |
| Blennow et al.19 | |
| Knight-Jones et al.20 | |
| Zenker et al.21 | |
Uno de los aspectos más comprometidos en el síndrome es el lenguaje. La intención comunicativa está limitada durante los primeros años y tiende a mejorar con el tiempo, con extensión a un repertorio de gestos. Se presentan dificultades importantes en la aparición del lenguaje expresivo, estando ausente o muy pobre en la mayoría de los individuos. La comprensión del lenguaje «parece» limitada a un contexto específico, logran comprender principalmente aspectos de su entorno inmediato o aspectos donde han recibido mucho entrenamiento. La capacidad de escritura se limita al garabateo, algunos niños reciben terapia destinada al aprendizaje de la escritura, pero no se han descrito avances en los mismos4,8,15,16,22,23.
En cuanto al desarrollo del área personal-social se han descrito alteraciones en aspectos de la socialización y de la conducta. Al nivel de la interacción algunos autores señalan presencia de rasgos autistas, como la falta de interés por las relaciones sociales y conductas estereotipadas y/o autoestimulantes, siendo la más común la exploración persistente de las manos; también pueden presentar aleteos, sacudir, golpear o balancear la cabeza y tendencia a oler, balancear o golpear objetos de manera repetitiva y sin sentido3,4,14,17–20.
A nivel de la conducta pueden observarse estallidos de mal humor y episodios de actividad física violenta, pudiendo golpear a otras personas o lanzar objetos. También pueden presentar comportamientos autolesivos (succión excesiva de los dedos, pellizcarse a sí mismos y morderse la mano/muñeca3,4,14,17,18,20).
Finalmente, dentro de los aspectos evolutivos se han descrito dificultades en el desarrollo de la conducta alimentaria. Dificultades tempranas para chupar, dificultades para alimentarse asociadas a hipotonía en fisuras orales y/o faciales, pobre coordinación para tragar con consecuente aspiración y/o reflujo gastroesofágico y vómito. En pocos sujetos se ha observado la presencia de estenosis hipertrófica del píloro9. En niños mayores se ha descrito hiperfagia en algunos4,14 casos y disfagia orofaríngea en otros9.
Características clínicas a nivel neurológico: convulsiones, alteraciones visuales y auditivas e hidrocefaliaLos estudios muestran que entre el 44% y el 58% de los niños diagnosticados con el síndrome presentan algún tipo de convulsión4,9,24.
En un estudio realizado con 60 pacientes4 el 44% (26) presentó convulsiones de inicio entre los 4 días y los 2 años y 8 meses de edad. Las crisis eran de diferentes tipos: clónicas, espasmos infantiles, parciales complejas, tónico/clónicas, ausencias atípicas, tónicas, o espasmos tónicos. El 25% (2) de los niños con convulsiones tenía espasmos infantiles de inicio entre los 3 y 10 meses de edad. En 14 niños los espasmos estaban asociados a un electroencefalograma hipsarrítmico, característica propia del síndrome de West. En 8 estaban precedidos por una convulsión clónica focal o generalizada que ocurría entre uno y 7 meses y medio antes. Los espasmos infantiles estaban bien controlados por corticotropina en 11 pacientes (73,3%), mientras que en 2 de ellos el síndrome de West evolucionó a una imagen electroclínica de tipo Lennox-Gastaut, con ausencias atípicas, espasmos tónicos y crisis tónicas nocturnas; uno de estos pacientes continuó siendo resistente a la medicación y otro evolucionó lentamente con los años4. Once pacientes del estudio tenían convulsiones variadas (clónica, tónica, tónico/clónica, parcial compleja), de inicio entre 2 semanas y 17 meses de edad, que estaban más controladas con medicamentos antiepilépticos de uso común (AED). En 21 pacientes las convulsiones pararon a los 6 años de edad y dejaron de recibir medicación durante algunos años.
En otro estudio realizado con 24 pacientes24 el 46% (11 niños) tenía epilepsia. El autor dividió el grupo entre los que tenían crisis médicamente intratables (3 de 11), los que tenían espasmos infantiles (3 de 11) y los que tenían epilepsia bien controlada con la medicación (5 de 11). Por otra parte, 3 (12,5%) de los 24 evaluados tenían una historia de 2 o menos convulsiones, que habían ocurrido al inicio de la infancia. En todos los pacientes con historia de convulsiones la descripción física dada por los padres incluía movimientos de tipo tónico o tónico/clónico de las extremidades concomitantes con desviación de los ojos en muchos casos. Todos los pacientes estaban siendo tratados con AED. Los pacientes que solo habían presentado crisis durante el inicio de la infancia recibieron AED durante un breve periodo durante el primer año de vida.
A nivel visual las alteraciones oftalmológicas más comunes asociadas al síndrome son estrabismo, nistagmus, errores refractivos e inatención visual4,9. En menor grado se pueden observar cataratas, astigmatismo, fotofobia, coloboma en el nervio óptico y albinismo retinal4,25. Algunas de estas afecciones se relacionan con alteraciones al nivel del nervio óptico y del nervio troclear, otras con anomalías del disco óptico. Cerca del 80% de los niños con este síndrome presentan dificultades de este tipo.
Por otra parte, cerca de 2/3 de los niños presentan dificultades a nivel auditivo. Pueden presentar pérdida auditiva muy variada (de leve a severa). La pérdida puede ser de tipo neurosensorial, causada por alteraciones del nervio auditivo o conductiva, causada por alteraciones del oído medio26.
Otra de las complicaciones neurológicas que se ha descrito (aunque con menos frecuencia) es la hidrocefalia. En 1995 se publicó la descripción clínica de 3 pacientes, 2 de los cuales la presentaban13. En un estudio posterior se detectaron anomalías cerebrales en la ecografía prenatal en 2 casos. En un caso se observó de moderada a severa hidrocefalia no obstructiva mediante imagen de resonancia magnética (IRM) posnatal, y en el otro caso, que se trataba de un feto, la autopsia demostró hidrocefalia. El síndrome en los 2 casos fue establecido posnatalmente27.
Hallazgos en el electroencefalograma y en la resonancia magnéticaDentro de los estudios del síndrome se encuentran hallazgos electroencefalográficos variados, tales como hipsarritmia, picos focales y multifocales, pico lento generalizado y ondas complejas, asimetría de actividad de ondas lentas, rápido ritmo de reclutamiento y actividad de fondo lenta18,20,24.
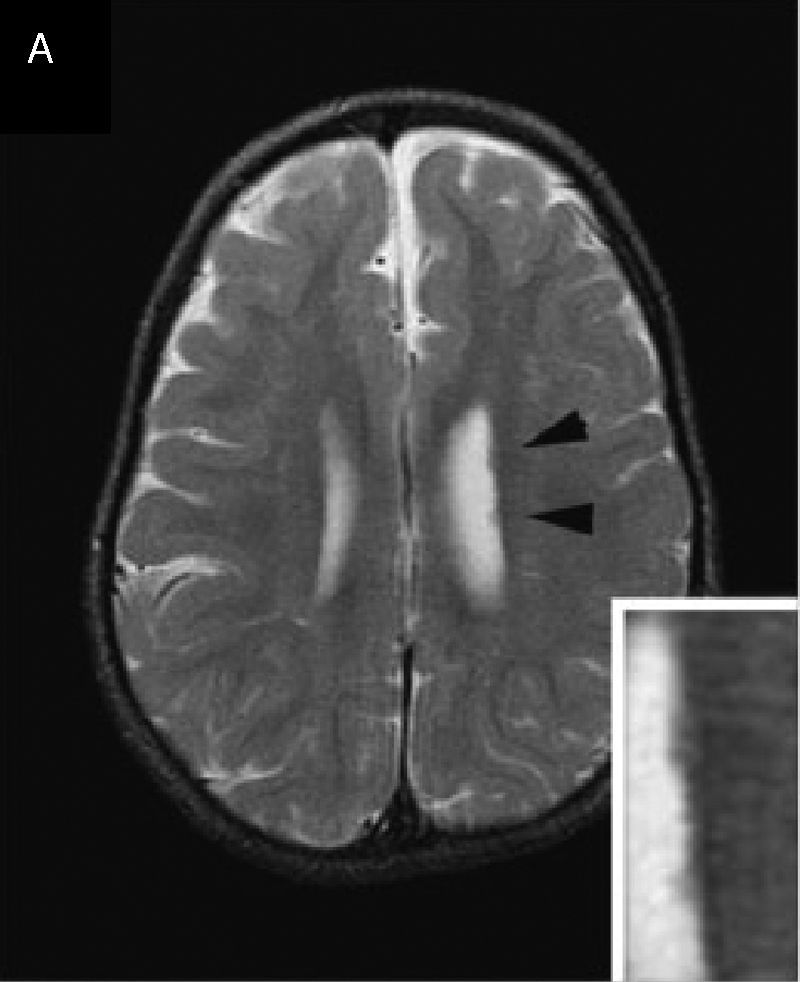
En cuanto a los hallazgos por IRM en 2006 se describió el caso de una paciente (niña de 3 años de edad) con cariotipo 1p36.22->1pter asociada a heterotopia periventricular28. En la imagen se observó heterotopia de varios nódulos de sustancia gris a lo largo de la pared del ventrículo izquierdo. El rostro del cuerpo calloso estaba truncado, los ventrículos estaban ligeramente alargados y se veían áreas irregulares de hiperintensidad en la sustancia blanca periventricular y subcortical consistente con mielinización tardía (fig. 2).

Heterotopia nodular periventricular. La figura corresponde a la resonancia magnética de un paciente con diagnóstico de síndrome de deleción 1p36.
Fuente: reproducido de Blennow et al.19 con autorización.
En un estudio posterior las IRM de 49 pacientes mostraron: ensanchamiento de los ventrículos laterales (en 18 pacientes), atrofia cortical (en 10 pacientes), ensanchamiento de espacios subaracnoideos (en 11 pacientes), atrofia cerebral difusa (en 5 pacientes) y ensanchamiento del opérculo frontotemporal (en 2 pacientes)4.
Dentro de este mismo estudio se observaron anomalías en la sustancia blanca en 8 pacientes, retraso en la mielinización (4 pacientes), áreas multifocales hiperintensas en imágenes T2 (en un paciente), leucomalacia periventricular (en 3 pacientes) y 2 pacientes tenían una malformación de Chiari tipo 1. Se encontraron también alteraciones de las estructuras comisurales (en 8 pacientes); hipoplasia, adelgazamiento y ensanchamiento total o parcial del cuerpo calloso (en 6 pacientes) y cavum septum pellucidum (en un paciente).
Son pocos los estudios que muestran hallazgos prenatales. En 2008 se publicaron hallazgos de la ecografía prenatal de 2 casos que fueron diagnosticados posnatalmente. El primer caso presentaba: ventriculomegalia, arteria umbilical única, malformaciones en un pie, defecto septal ventricular y retraso en el crecimiento intrauterino. La IRM posnatal de este caso mostraba hidrocefalia no obstructiva moderada-severa, colpocefalia y mielinización anormal de la extremidad o brazo anterior de la cápsula interna27. El segundo caso era un feto en el que se observaron alteraciones relacionadas con holoprosencefalia en la ecografía prenatal. La autopsia fetal demostró hidrocefalia, polimicrogiria focal e hipoplasia cerebelosa27.
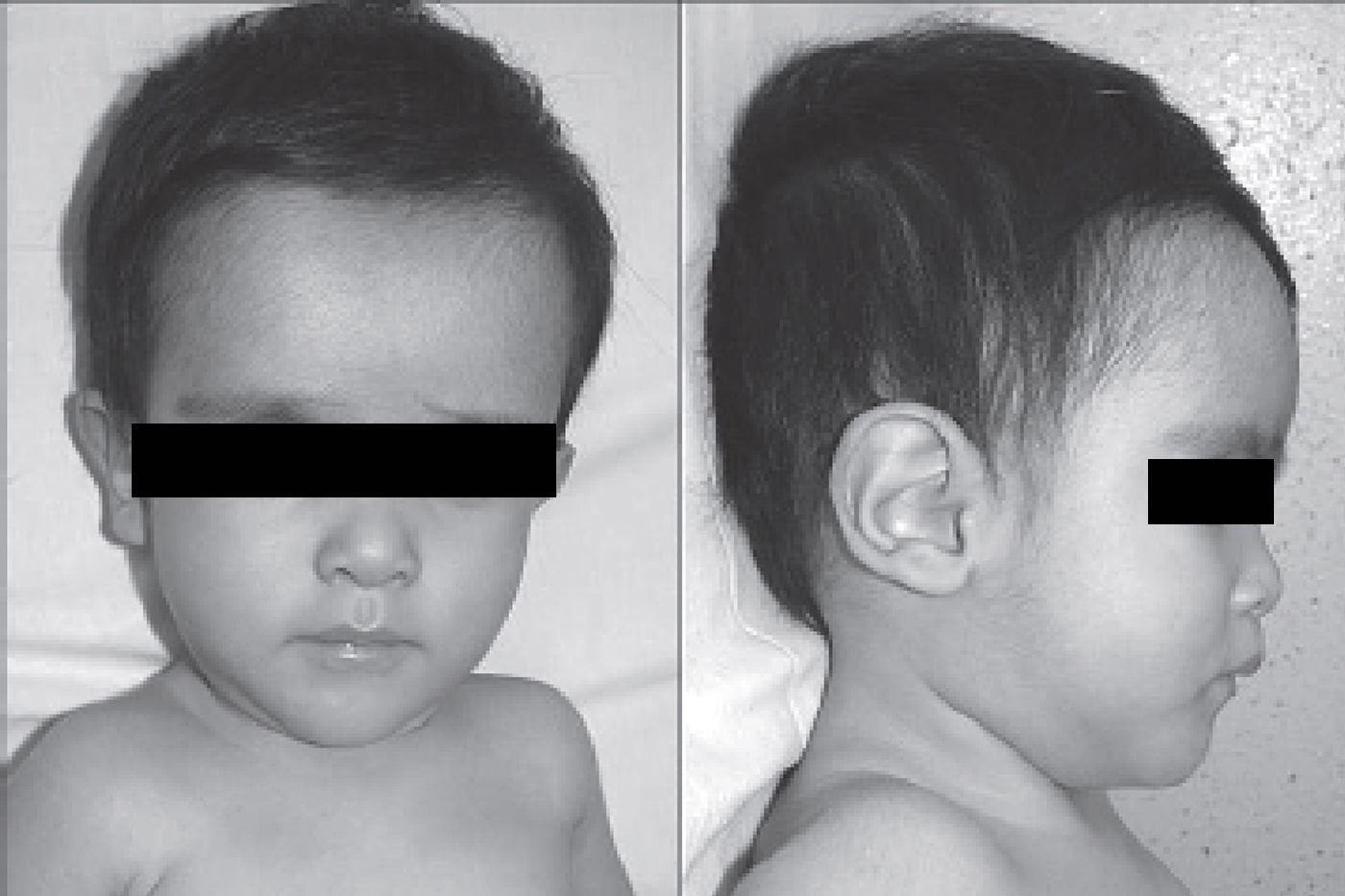
Características clínicas a nivel de rasgos craneofaciales dismórficosDentro del síndrome se encuentran los siguientes rasgos: microbraquicefalia (65%), fontanela anterior grande y con cierre tardío (77%), cejas rectas (100%), ojos hundidos (100%), epicanto (50%), raíz/puente nasal anchos (100%), hipoplasia del tercio medio facial (100%), orejas implantadas anormalmente (posteriormente rotadas/bajas) (40%), filtrum largo (100%), barbilla puntiaguda (100%)4. Adicionalmente se han observado hélices de los oídos engrosadas3,14. Algunas características mencionadas pueden observarse en la figura 3. Entre el 10% y el 40% de pacientes descritos presentan fisura orofacial, y en casos aislados se ha llegado a observar presencia de fisura labiopalatina, paladar ojival, fisura submucosa del paladar y crestas palatinas prominentes22.

Características craneofaciales. La figura presenta fotografías frontales y laterales de pacientes diagnosticados con el síndrome de deleción 1p36, en las que se pueden apreciar algunos de los rasgos craneofaciales propios del síndrome.
Fuente: tomado de Villarroel et al.5 con autorización.
Las alteraciones a nivel cardiaco ocurren entre el 43% y el 71% de los pacientes e incluyen: defectos septales auriculares y ventriculares, anomalías valvulares, ductus arterioso persistente, tetralogía de Fallot, coartación de la aorta, estenosis o estrechamiento infundibular del ventrículo derecho y anomalía de Ebstein. El 27% presentan miocardiopatía en los primeros años de vida. En el 23% esta miocardiopatía es de tipo no compactada, y tiende a mejorar con el tiempo4,9.
Características clínicas a nivel esqueléticoSe presentan malformaciones esqueléticas aproximadamente en el 40% de los pacientes e incluyen: retraso en el crecimiento óseo, braquidactilia (por ejemplo pies y manos cortos), escoliosis, anomalías en las costillas y extremidades inferiores asimétricas8. En algunos pacientes también se ha observado estenosis espinal congénita y clinodactilia14,17. En muy pocos pacientes se observó polidactilia13. En algunos estudios se ha descrito la presencia de microcefalia3. Algunas de las características a nivel de las manos se pueden observar en la figura 4.

Características esqueléticas. En las fotografías se puede apreciar algunas de las características a nivel esquelético, como braquidactilia y clinodactilia del quinto dedo.
Fuente: modificado de Battaglia et al.4.
Las anomalías genitourinarias pueden presentarse aproximadamente en el 22% de los individuos e incluyen: pelvis renal unilateral con hidronefrosis del polo superior, ectopia renal con quiste en el riñón derecho y ectasia pélvica. En una minoría de hombres se ha descrito criptorquidia, hipospadias, hipoplasia escrotal y micropenes. Y, en mujeres, se han descrito labios menores pequeños y clítoris pequeño, labios mayores hipertróficos e hipoplasia uterina4.
Características clínicas a nivel endocrinológicoSe ha descrito hipotiroidismo en el 15-20% de individuos de varias edades que presentan el síndrome4,9.
Características clínicas a nivel dermatológicoAlteraciones a este nivel se han descrito en pocos individuos, a saber: arañas vasculares en la piel por dilatación de capilares pequeños (telangiectasia), máculas hiperpigmentadas13 y excesiva piel en el cuello29.
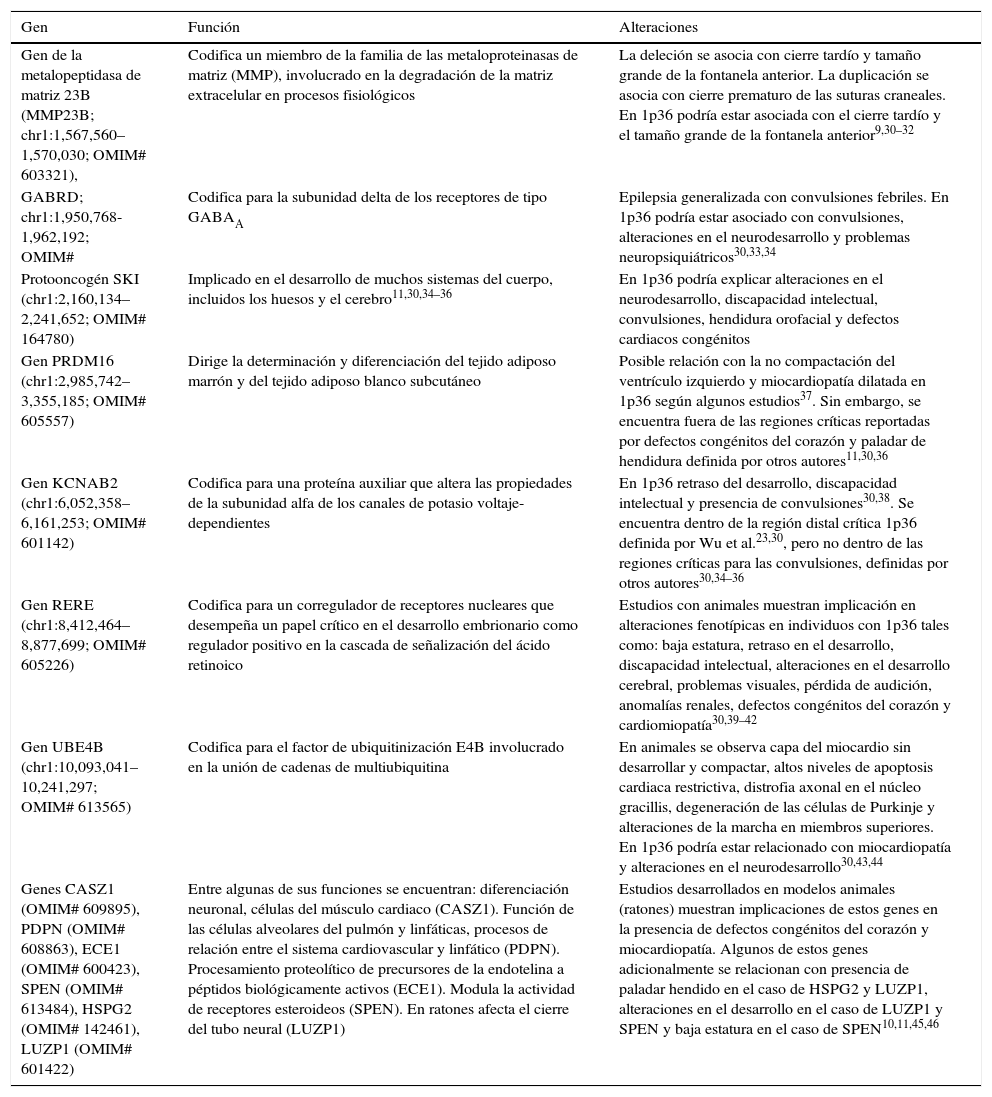
Genes responsables implicados en las características fenotípicas del síndrome de deleción (monosomía) 1p36Una completa actualización sobre los genes implicados en algunas características de este síndrome ha sido realizada recientemente (2015) por Jordan et al.30. Los estudios revisados en algunos casos se basan en hallazgos obtenidos con pacientes y en otros casos en hallazgos obtenidos con modelos animales. Los genes mencionados implicados en algunas de las características del síndrome 1p36 se indican en la tabla 2.
Genes responsables implicados en las características fenotípicas de 1p36
| Gen | Función | Alteraciones |
|---|---|---|
| Gen de la metalopeptidasa de matriz 23B (MMP23B; chr1:1,567,560–1,570,030; OMIM# 603321), | Codifica un miembro de la familia de las metaloproteinasas de matriz (MMP), involucrado en la degradación de la matriz extracelular en procesos fisiológicos | La deleción se asocia con cierre tardío y tamaño grande de la fontanela anterior. La duplicación se asocia con cierre prematuro de las suturas craneales. En 1p36 podría estar asociada con el cierre tardío y el tamaño grande de la fontanela anterior9,30–32 |
| GABRD; chr1:1,950,768-1,962,192; OMIM# | Codifica para la subunidad delta de los receptores de tipo GABAA | Epilepsia generalizada con convulsiones febriles. En 1p36 podría estar asociado con convulsiones, alteraciones en el neurodesarrollo y problemas neuropsiquiátricos30,33,34 |
| Protooncogén SKI (chr1:2,160,134–2,241,652; OMIM# 164780) | Implicado en el desarrollo de muchos sistemas del cuerpo, incluidos los huesos y el cerebro11,30,34–36 | En 1p36 podría explicar alteraciones en el neurodesarrollo, discapacidad intelectual, convulsiones, hendidura orofacial y defectos cardiacos congénitos |
| Gen PRDM16 (chr1:2,985,742–3,355,185; OMIM# 605557) | Dirige la determinación y diferenciación del tejido adiposo marrón y del tejido adiposo blanco subcutáneo | Posible relación con la no compactación del ventrículo izquierdo y miocardiopatía dilatada en 1p36 según algunos estudios37. Sin embargo, se encuentra fuera de las regiones críticas reportadas por defectos congénitos del corazón y paladar de hendidura definida por otros autores11,30,36 |
| Gen KCNAB2 (chr1:6,052,358–6,161,253; OMIM# 601142) | Codifica para una proteína auxiliar que altera las propiedades de la subunidad alfa de los canales de potasio voltaje-dependientes | En 1p36 retraso del desarrollo, discapacidad intelectual y presencia de convulsiones30,38. Se encuentra dentro de la región distal crítica 1p36 definida por Wu et al.23,30, pero no dentro de las regiones críticas para las convulsiones, definidas por otros autores30,34–36 |
| Gen RERE (chr1:8,412,464–8,877,699; OMIM# 605226) | Codifica para un corregulador de receptores nucleares que desempeña un papel crítico en el desarrollo embrionario como regulador positivo en la cascada de señalización del ácido retinoico | Estudios con animales muestran implicación en alteraciones fenotípicas en individuos con 1p36 tales como: baja estatura, retraso en el desarrollo, discapacidad intelectual, alteraciones en el desarrollo cerebral, problemas visuales, pérdida de audición, anomalías renales, defectos congénitos del corazón y cardiomiopatía30,39–42 |
| Gen UBE4B (chr1:10,093,041–10,241,297; OMIM# 613565) | Codifica para el factor de ubiquitinización E4B involucrado en la unión de cadenas de multiubiquitina | En animales se observa capa del miocardio sin desarrollar y compactar, altos niveles de apoptosis cardiaca restrictiva, distrofia axonal en el núcleo gracillis, degeneración de las células de Purkinje y alteraciones de la marcha en miembros superiores. En 1p36 podría estar relacionado con miocardiopatía y alteraciones en el neurodesarrollo30,43,44 |
| Genes CASZ1 (OMIM# 609895), PDPN (OMIM# 608863), ECE1 (OMIM# 600423), SPEN (OMIM# 613484), HSPG2 (OMIM# 142461), LUZP1 (OMIM# 601422) | Entre algunas de sus funciones se encuentran: diferenciación neuronal, células del músculo cardiaco (CASZ1). Función de las células alveolares del pulmón y linfáticas, procesos de relación entre el sistema cardiovascular y linfático (PDPN). Procesamiento proteolítico de precursores de la endotelina a péptidos biológicamente activos (ECE1). Modula la actividad de receptores esteroideos (SPEN). En ratones afecta el cierre del tubo neural (LUZP1) | Estudios desarrollados en modelos animales (ratones) muestran implicaciones de estos genes en la presencia de defectos congénitos del corazón y miocardiopatía. Algunos de estos genes adicionalmente se relacionan con presencia de paladar hendido en el caso de HSPG2 y LUZP1, alteraciones en el desarrollo en el caso de LUZP1 y SPEN y baja estatura en el caso de SPEN10,11,45,46 |
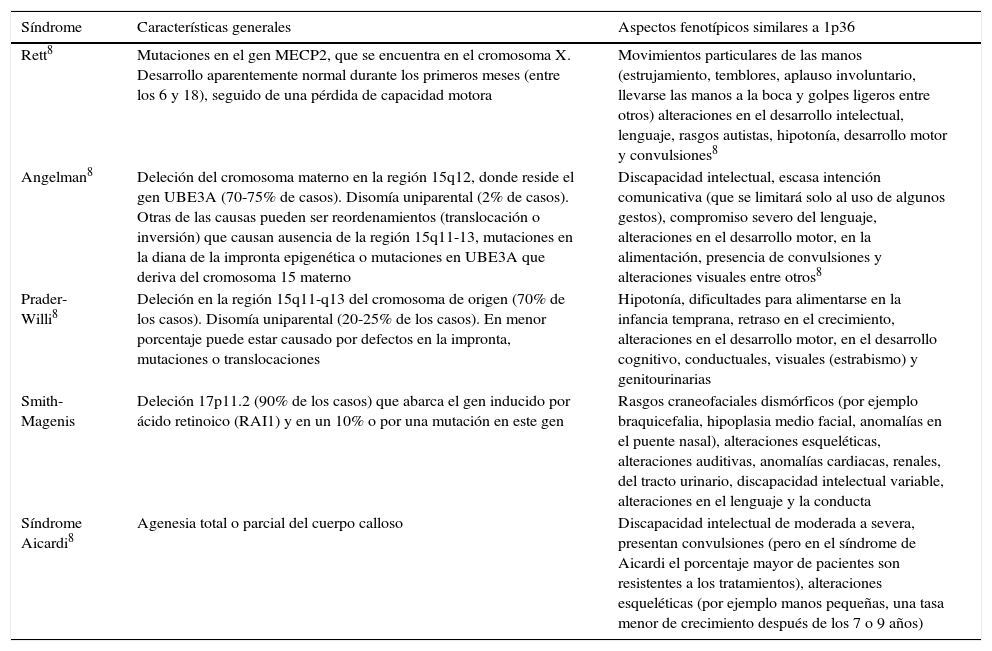
Battaglia8 sugiere que en ocasiones puede realizarse un mal diagnóstico debido a que algunos síntomas se solapan o superponen a los de otros síndromes. En la tabla 3 se mencionan los diagnósticos diferenciales.
Diagnósticos diferenciales
| Síndrome | Características generales | Aspectos fenotípicos similares a 1p36 |
|---|---|---|
| Rett8 | Mutaciones en el gen MECP2, que se encuentra en el cromosoma X. Desarrollo aparentemente normal durante los primeros meses (entre los 6 y 18), seguido de una pérdida de capacidad motora | Movimientos particulares de las manos (estrujamiento, temblores, aplauso involuntario, llevarse las manos a la boca y golpes ligeros entre otros) alteraciones en el desarrollo intelectual, lenguaje, rasgos autistas, hipotonía, desarrollo motor y convulsiones8 |
| Angelman8 | Deleción del cromosoma materno en la región 15q12, donde reside el gen UBE3A (70-75% de casos). Disomía uniparental (2% de casos). Otras de las causas pueden ser reordenamientos (translocación o inversión) que causan ausencia de la región 15q11-13, mutaciones en la diana de la impronta epigenética o mutaciones en UBE3A que deriva del cromosoma 15 materno | Discapacidad intelectual, escasa intención comunicativa (que se limitará solo al uso de algunos gestos), compromiso severo del lenguaje, alteraciones en el desarrollo motor, en la alimentación, presencia de convulsiones y alteraciones visuales entre otros8 |
| Prader-Willi8 | Deleción en la región 15q11-q13 del cromosoma de origen (70% de los casos). Disomía uniparental (20-25% de los casos). En menor porcentaje puede estar causado por defectos en la impronta, mutaciones o translocaciones | Hipotonía, dificultades para alimentarse en la infancia temprana, retraso en el crecimiento, alteraciones en el desarrollo motor, en el desarrollo cognitivo, conductuales, visuales (estrabismo) y genitourinarias |
| Smith-Magenis | Deleción 17p11.2 (90% de los casos) que abarca el gen inducido por ácido retinoico (RAI1) y en un 10% o por una mutación en este gen | Rasgos craneofaciales dismórficos (por ejemplo braquicefalia, hipoplasia medio facial, anomalías en el puente nasal), alteraciones esqueléticas, alteraciones auditivas, anomalías cardiacas, renales, del tracto urinario, discapacidad intelectual variable, alteraciones en el lenguaje y la conducta |
| Síndrome Aicardi8 | Agenesia total o parcial del cuerpo calloso | Discapacidad intelectual de moderada a severa, presentan convulsiones (pero en el síndrome de Aicardi el porcentaje mayor de pacientes son resistentes a los tratamientos), alteraciones esqueléticas (por ejemplo manos pequeñas, una tasa menor de crecimiento después de los 7 o 9 años) |
El tratamiento de pacientes con el síndrome 1p36 es multidisciplinario. El manejo de las dificultades en el neurodesarrollo implica fisioterapia para las alteraciones motoras, musculares y esqueléticas. Algunos niños con alteraciones esqueléticas graves requerirán cirugía8. La terapia de lenguaje trabajará sobre déficits en la comunicación, el lenguaje y el habla, aspectos relacionados con la deglución-ingesta e hipotonía facial. Algunos pacientes con paladar hendido requerirán de técnicas e instrumentos especiales para la ingesta (como el Haberman feeder) y el uso de sondas gástricas en casos más graves8.
Se requerirá manejo neuropsicológico para las dificultades cognitivas a nivel de atención, memoria, percepción, funciones ejecutivas y aprendizaje, así como manejo psicológico para las alteraciones de la conducta, la interacción social y la conducta adaptativa. También es imprescindible del trabajo de educadores especiales o psicopedagogos en relación con aspectos curriculares escolares y con mecanismos de integración escolar.
No existen hasta la fecha publicaciones que establezcan mecanismos específicos de tratamiento de alteraciones en el neurodesarrollo, lo que hace difícil establecer pautas de manejo que guíen a los distintos profesionales.
A nivel farmacológico8, para el manejo de crisis epilépticas se plantea que todos los tipos de convulsiones en estos pacientes pueden ser controlados por AED, que deben ser administrados tan pronto como sea posible. Los defectos congénitos del corazón también han de ser tratados con fármacos estándar. Las dificultades de inatención visual se podrán manejar con un programa de rehabilitación apropiado. Los estrabismos se podrán mejorar con cirugías de corrección. La pérdida auditiva se trata con pruebas de audífonos.
ConclusionesLos pacientes con síndrome 1p36 presentan alteraciones al nivel de varios sistemas y cursan con compromiso del desarrollo evolutivo global. Aunque en la actualidad existe una intensa actividad investigadora sobre este síndrome y los distintos genes responsables involucrados en el mismo, todavía resta conseguir la determinación de todos los que contribuyen a ser un factor de riesgo que permitan en el futuro su prevención y potencialmente su futuro tratamiento. Por el momento, no existe tratamiento médico efectivo y se debe abordar utilizando fármacos que palien algunos de los síntomas asociados y mediante tratamiento neuropsicológico adecuado.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónMinisterio de Economía y Competitividad, España. Grant BFU2012-38208.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
