


La variante de Dandy Walker se define como una hipoplasia variable del vermis cerebeloso, con o sin aumento de la fosa posterior y sin elevación del tentorio.
ObjetivoDescribir el caso de una enfermedad poco frecuente y hacer énfasis en la necesidad de precisar la etiología de malformaciones prenatales en niños que son clasificados erróneamente como parálisis cerebral secundaria a asfixia, así como su manejo multidisciplinario.
Caso clínicoPaciente varón, de 8 años de edad, con antecedentes de parálisis cerebral infantil, epilepsia y retraso del desarrollo, que fue ingresado por historia de convulsiones tónico-clónicas. Durante su hospitalización presentó múltiples episodios convulsivos, controlados con anticonvulsivantes. Se realizó tomografía computarizada, observándose comunicación entre la cisterna magna y el cuarto ventrículo; este último aumentado de tamaño. Además, el vermis del cerebelo presentaba hipoplasia parcial, siendo estos hallazgos compatibles con una variante del síndrome Dandy Walker.
ConclusiónLa variante de Dandy Walker puede ser sintomática o asintomática, y las imágenes encontradas no necesariamente se relacionan con las alteraciones del desarrollo, debido a los múltiples síndromes y alteraciones cromosómicas vinculadas a este cuadro. La presentación clínica y el pronóstico dependen de las alteraciones presentes. Por ello, es importante un manejo multidisciplinario considerando que el tratamiento depende de los síntomas presentados.
Dandy Walker variant is defined by a variable hypoplasia of the cerebellar vermix with or without posterior fossa increase and without tentorium elevation.
Objectivedescribe the case of a rare disease and emphasise the need to clarify the aetiology of prenatal malformations, as well as its multidisciplinary management.
Case reportA male patient, 8 years of age, with a history of Infantile Cerebral Palsy and epilepsy, who was admitted with a history of tonic-clonic seizures. He was admitted due to psycho-motor developmental delay. During his hospitalisation, he had multiple seizure episodes, controlled with anticonvulsants. A computerized tomography was performed, in which communication was observed between the cisterna magna and fourth ventricle (the latter increased in size). In addition, the cerebellar vermix showed a partial hypoplasia. All these findings were compatible with a variant of the Dandy Walker syndrome.
ConclusionDandy Walker variant may be asymptomatic and the images found may not indicate them as the cause of developmental disorders, due to its association with multiple syndromes and chromosomal abnormalities. Clinical presentation and prognosis depends on the related disorders, and a multidisciplinary approach is important, because the treatment depends on the symptoms presented.
El complejo Dandy Walker tiene una prevalencia de uno por cada 35.000 nacidos vivos en Estados Unidos, y se refiere a un conjunto de anomalías del sistema nervioso central (SNC), más específicamente de la fosa posterior constituida por 3 cuadros imagenológicos1,2: 1) malformación de Dandy Walker; 2) variante de Dandy Walker; y 3) mega cisterna magna. La variante de Dandy Walker se define como una hipoplasia variable del vermis cerebeloso con o sin aumento de la fosa posterior, y sin elevación del tentorio. Algunos autores recomiendan usar el término hipoplasia aislada del vermis cerebeloso en lugar de variante de Dandy Walker2.
Las anomalías cromosómicas se asocian entre el 17,6% y el 54%2, siendo la variante Dandy Walker parte de más de 50 síndromes genéticos, altamente relacionados con alteraciones en el cariotipo3. El porcentaje de anomalías cromosómicas se incrementa mucho en los casos diagnosticados de forma temprana, por lo que es de especial importancia la genealogía familiar de al menos 3 generaciones, con particular atención en casos de retardo mental, retraso en el desarrollo, malformaciones congénitas, abortos, mortinatos y muerte infantil que oriente a un modo de herencia y permita evaluar enfermedades con expresividad variable4,5.
Las malformaciones asociadas del SNC o fuera de él se presentan entre el 50% y el 70% de los casos. Las más frecuentes son las del SNC, y se pueden corresponder a: ventriculomegalia, encefalocele, holoprosencefalia, microcefalia, malformaciones de las circunvoluciones cerebrales2,4 y defectos de la línea media, como agenesia del cuerpo calloso3 (malformación reportada en una de 19.000 autopsias)6. De las anormalidades sistémicas la polidactilia y los defectos cardiacos son los más comunes7.
El cuadro clínico es inespecífico, y depende de las alteraciones cerebrales. Las manifestaciones clínicas pueden corresponder a lentitud del desarrollo psicomotor, aumento progresivo del perímetro cefálico y fontanela tensa en caso de lactantes cuando existe aumento de presión intracraneal. El tamaño del vermis influye tanto en el desarrollo neurológico como en el de la inteligencia. En preescolares y adolescentes se reporta una frecuencia de déficit en la inteligencia del 40-70%3. Puede existir signos de disfunción cerebelosa4, retraso en el desarrollo motor, en la expresión verbal, hipotonía en la musculatura axial2, vómitos, irritabilidad, cefalea, convulsiones y papiledema1.
Las técnicas diagnósticas por imagen incluyen: ultrasonido, resonancia magnética (RM) y tomografía computarizada (TC). La evaluación inicial debe hacerse con ultrasonido, que es el método de elección para el diagnóstico prenatal, siendo posible vía transvaginal un diagnóstico a partir de la semana 18 de gestación8. En la actualidad la RM es considerada la herramienta diagnóstica más precisa para el diagnóstico de Dandy Walker y sus variantes8.
El tratamiento consiste en tratar los problemas asociados4. El pronóstico es variable y depende de las malformaciones relacionadas. Se ha descrito supervivencia del 15% de los pacientes que fueron diagnosticados en el embarazo y con síntomas en edad temprana4. El síndrome de Dandy Walker como anormalidad aislada tiene alta posibilidad de supervivencia, y hay reportes de personas que han tenido este diagnóstico durante toda su vida sin ningún síntoma3. Mientras más temprano se inician de síntomas mayor es la mortalidad. El género, la raza y el ingreso monetario no son predictores significativos en la mortalidad9.
El objetivo de este artículo es describir el caso de una enfermedad poco frecuente en los pacientes pediátricos y, a la vez, hacer énfasis en la necesidad de investigar y precisar la etiología de malformaciones prenatales en niños que son clasificados erróneamente como parálisis cerebral secundaria a asfixia, así como su manejo multidisciplinario.
Caso clínicoPaciente varón, de 8 años de edad, con los diagnósticos de parálisis cerebral infantil y epilepsia secundaria a asfixia neonatal. Se ingresó con historia de episodios convulsivos tónico clónicos en 3 ocasiones acompañados de sialorrea, versión ocular y relajación de esfínter vesical. El paciente había sido ingresado previamente por un cuadro similar, donde se indicó tratamiento con ácido valproico y fenitoína, con los cuales, según su madre, hubo mejoría clínica. Las crisis convulsivas comenzaron a los 7 meses de edad.
En sus antecedentes personales destacaba un soplo cardiaco detectado al nacimiento, evaluado por un cardiólogo pediatra, quien descartó la presencia de cardiopatía.
La madre no relataba antecedentes familiares de retraso mental o epilepsia. Tenía historia de un aborto, un óbito, un hijo muerto el cual falleció al nacer por asfixia y un hijo vivo (el caso clínico) y embarazo actual. Cada embarazo de diferente padre.
Al examen físico de ingreso destacaba un paciente severamente desnutrido con peso de 15kg (menor al percentil 3 peso para la edad, según tablas del Center for Disease Control and Prevention [CDC]), talla 106cm (menor al percentil 3 talla para la edad, tablas CDC) y con índice de masa corporal 13,349 (menor al percentil 3, tablas CDC). Buen estado general, frecuencia cardiaca 125 latidos por minuto, frecuencia respiratoria de 19 respiraciones por minuto y temperatura de 37 grados centígrados, presentando retraso del desarrollo motor y retraso del habla. Intentaba comunicación con gestos, expresión facial y señalando, no caminaba, ni se alimentaba solo, no presentaba atrofia muscular ni parálisis de los nervios faciales.
Durante su primer día de hospitalización presentó 4 episodios convulsivos tónico-clónicos, con relajación de esfínteres que fueron controlados con la administración de diazepam. Luego se realizó impregnación con fenitoína y se continuó con un plan de vigilancia para realizar estudios. El segundo y tercer día el paciente continuó presentando crisis. Se realizaron niveles plasmáticos de fenitoina y ácido valproico que reportaron 16,8μg/dl (valor de referencia 10-20μg/dl) y ≤10μg/dl (valor de referencia 50-100μg/dl) respectivamente y se ajustó la dosis de ácido valproico.
Durante el cuarto día permaneció estable, sin presentar nuevos episodios convulsivos, por lo que tras vigilancia por 24h adicionales fue dado de alta con indicación de seguimiento y control por neurología pediátrica.
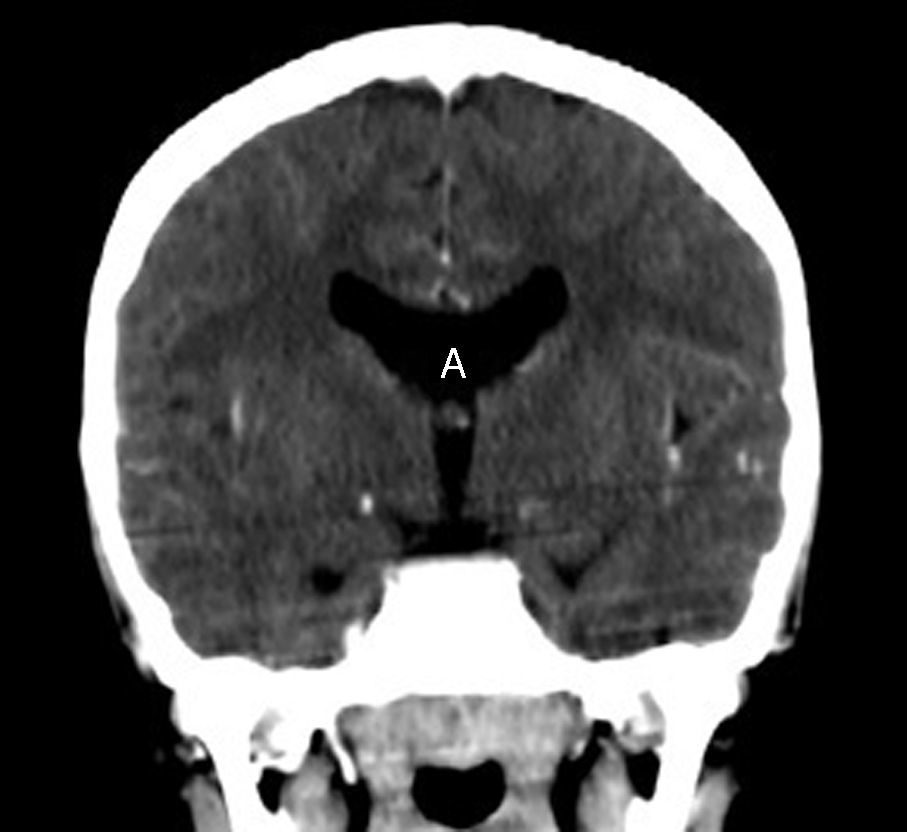
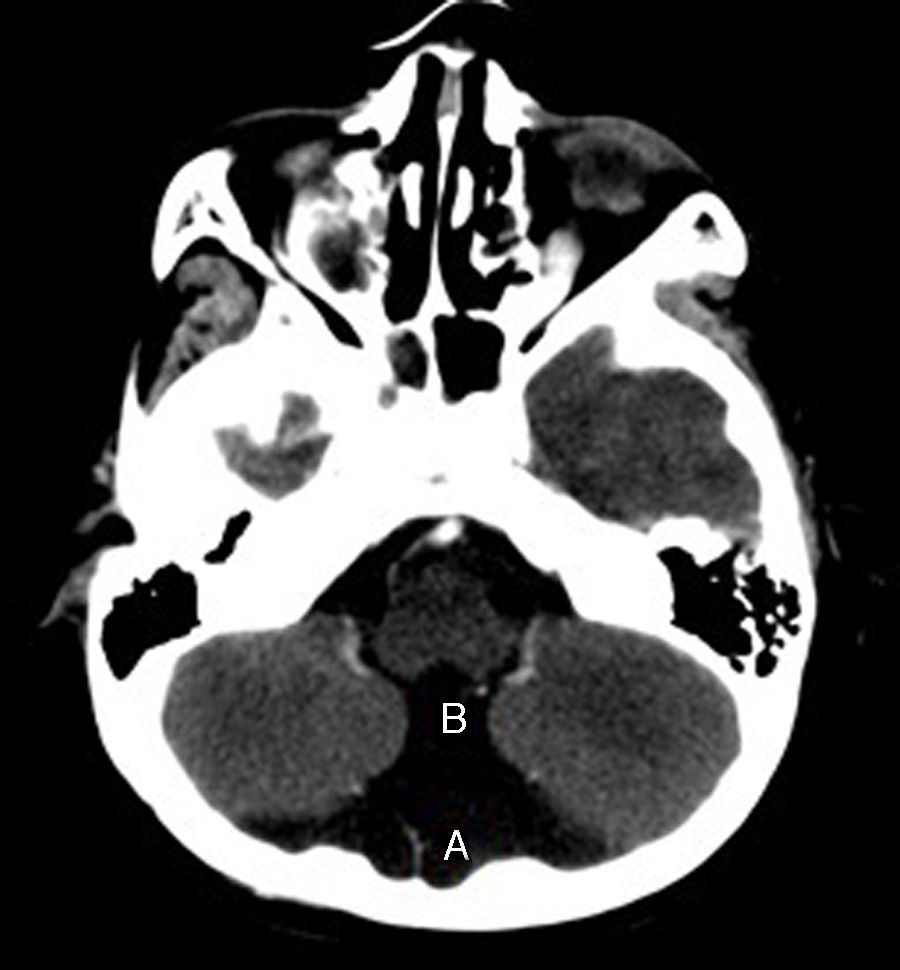
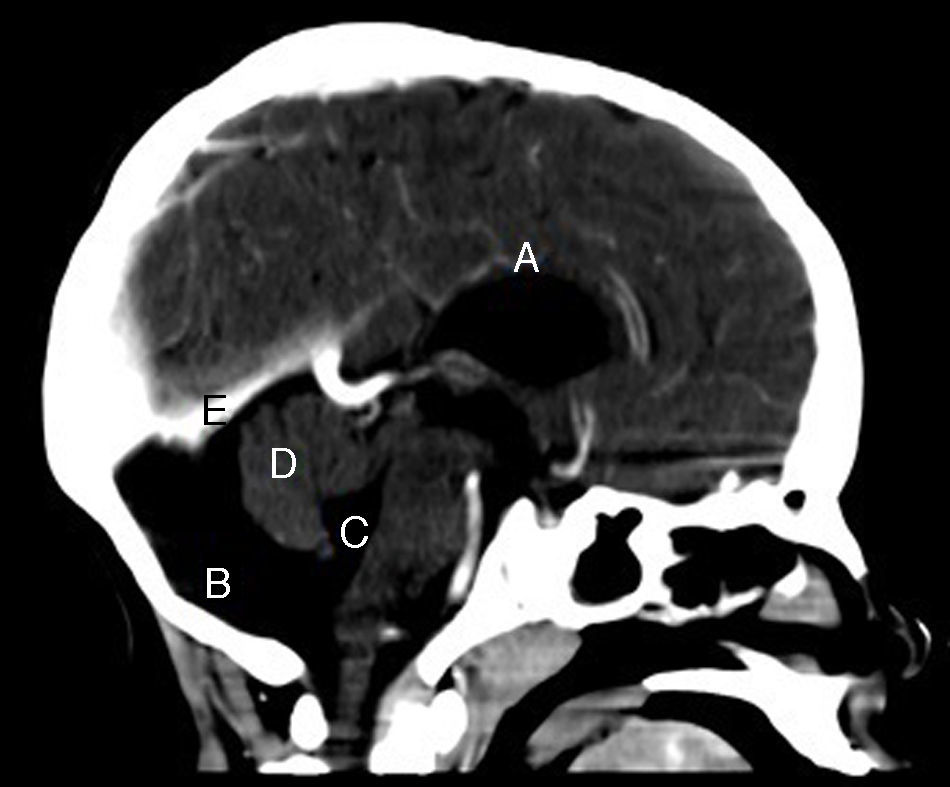
En el control neurológico se realizó TC, donde se constató ausencia de septum pellucidum y comunicación entre la cisterna magna y el cuarto ventrículo, este último aumentado de tamaño (figs. 1 y 2). Se visualizó, además, un vermis cerebeloso con hipoplasia parcial y una agenesia del cuerpo calloso (fig. 3), hallazgos compatibles con una variante del síndrome de Dandy Walker, lo que permitió el diagnóstico.
.")
 y el cuarto ventrículo aumentado de tamaño (B).")
, fosa posterior normal (B), comunicación con el cuarto ventrículo levemente aumentado de tamaño (C), vermis cerebeloso que muestra hipoplasia parcial (D), sin elevación del tentorio (E).")
El paciente continuó con su tratamiento anticonvulsivante y evaluación por neurología pediátrica. A los 6 meses de seguimiento no volvió a presentar crisis convulsivas; también continuó en manejo por un nutricionista y un gastroenterólogo pediatra para mejorar su estado nutricional.
DiscusiónLa literatura reporta una incidencia de variante de Dandy Walker de uno en 25 a 35.000 recién nacidos vivos, con predominio en mujeres sobre hombres (en relación 3:1), con una mortalidad general, independiente de la sintomatología o de la edad al diagnóstico, aproximada del 5% al 20%8,10. En ocasiones puede haber subdiagnóstico; esto se debe a que se adjudica la etiología de las dificultades neurológicas erróneamente a asfixia neonatal, como en el caso presentado, y en realidad se trata de una malformación prenatal que puede ser de diversas etiologías y que requiere que se investigue la causa genética.
La mayoría de los afectados presenta síntomas durante el primer año de vida, los que se pueden manifestar con retraso en el desarrollo psicomotor, dificultades visuales y auditivas, hipotonía, anomalías en la respiración, espasticidad y convulsiones, mientras que otros pueden no presentar manifestaciones clínicas. Se han detectado de manera incidental personas asintomáticas en la edad adulta, en estudio por motivos ajenos al síndrome11,12. En nuestro paciente la clínica concuerda con la literatura consultada, que refiere que aproximadamente el 90% presenta síntomas en el primer año de vida13.
La etiología de la variante de Dandy Walker aún no está claramente definida y se ha sugerido predisposición genética. Ha sido identificada en múltiples síndromes y anomalías cromosómicas7, generalmente asociada a las trisomías 13, 18 y 21, 3q, 6p, tetrasomía 9p, deleción 13q, así como translocaciones cromosómicas14, procesos infecciosos prenatales como TORCH y exposición a ciertos agentes químicos como alcohol y warfarina8,11. Por esta razón, la sola presencia de malformaciones en estudios de imagen no puede ser relacionada como la única causa de alteraciones neurológicas en el paciente.
Se ha estimado que el 40% de los casos de retraso en el desarrollo son de causa genética5. En este caso los antecedentes gineco-obstétricos de la madre son interesantes, ya que relatan un aborto, un óbito y un hijo fallecido al nacer por asfixia neonatal, previos al nacimiento del paciente. No existen antecedentes familiares de retardo mental por parte materna, aunque se desconocen los antecedentes paternos. Esta situación podría estar relacionada con algún patrón de herencia.
La presentación clínica coincide con estudios que reportan que estos pacientes presentan convulsiones, dificultades visuales y auditivas, anomalías sistémicas, principalmente cardiológicas y otras anomalías del SNC, asociadas con pobre desarrollo intelectual7. Nuestro paciente presentó un soplo cardiaco neonatal, pero se descartó cardiopatía.
Los estudios de imagen realizados nos confirman el diagnóstico. La RM es el método de elección10; sin embargo, estos estudios no están al alcance de la mayoría de los centros13. El estudio mediante TC previa introducción de contraste hidrosoluble es muy útil para determinar los compartimentos de las lesiones quísticas13.
Variante Dandy Walker es un término que se ha utilizado para referirse a hipoplasia de vermis cerebeloso y un agrandamiento de la cisterna magna, sin aumento de la fosa posterior7. El cuarto ventrículo también está aumentado de tamaño y hay comunicación libre entre este y la cisterna de la base, además de dilatación del foramen de Magendie13,14.
El tamaño disminuido de los hemisferios cerebelosos está asociado a bajo intelecto7. El cerebelo es conocido por su rol en las funciones motoras y de coordinación, pero se ha relacionado también con aspectos del humor y la cognición15.
También es importante identificar anomalías supratentoriales asociadas, ya que el pronóstico de estos pacientes es mucho mejor en la ausencia de estas7. Es frecuente encontrar defectos de línea media asociados a disgenesia del cuerpo calloso en un 20% a 25% de los casos reportados.
Otro hallazgo en nuestro paciente fue la ausencia de septum pellucidum12; se puede observar elevación de la tórcula y el tentorio, pero estas alteraciones son consecuencia de la malformación principalmente encontradas en Dandy Walker y no en su variante8. Al examen microscópico de muestras de autopsia se han detectado alteraciones morfológicas importantes en neuronas y dendritas como gliosis, pobre arborización de dendritas y reducción del tamaño celular14.
El tratamiento se dirige a los síntomas, y si el paciente no los presenta no se indica tratamiento. La aparición de síntomas en la edad adulta es extremadamente rara, pero pueden presentarse espontáneamente o tras un traumatismo craneal16.
Es importante el abordaje integral de estos pacientes, dependiendo de las alteraciones asociadas que presenten, como derivación del ventrículo peritoneal en caso de hipertensión endocraneal16. Cuando se asocia a psicosis se indica tratamiento con antipsicóticos11. Resulta importante su manejo en un programa de rehabilitación con terapia de leguaje y ejercicios de psicomotricidad en los pacientes con alteraciones en el desarrollo motor y del habla8,12. En nuestro paciente persistieron las crisis convulsivas a pesar de estar con anticonvulsivantes, pero al corregir las dosis de ácido valproico se estabilizó su epilepsia. Es importante disponer de un equipo multidisciplinario para su estudio y manejo3. El pronóstico depende en gran medida de malformaciones asociadas.
La interrupción precoz del embarazo, cuando existe un diagnóstico certero, es una opción en algunos países17.
ConclusiónLa variante de Dandy Walker es la presencia de disgenesia o agenesia del vermis cerebeloso, dilatación del cuarto ventrículo y aumento del tamaño de la cisterna magna sin aumento de tamaño de la fosa posterior.
Es conveniente contar con exámenes de imagen cerebral en niños con significativos trastornos de su desarrollo psicomotor, incluso cuando tiene antecedentes de asfixia al nacer, aunque las imágenes encontradas no siempre se pueden señalar como causa del alteraciones del desarrollo, debido a su relación con múltiples síndromes y alteraciones cromosómicas.
La presentación clínica y el pronóstico dependen de las alteraciones relacionadas dentro y fuera del sistema nervioso central. El tamaño del cerebelo se suele asociar con el desarrollo intelectual, por lo que a mayor disgenesia menor es el desarrollo intelectual.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
