


Analizar la metilación en los promotores de los genes CDKN2B y DBC1 en muestras de pacientes con leucemia linfoblástica aguda (LLA), leucemia mieloblástica aguda (LMA) y leucemia mieloide crónica (LMC). Además, correlacionar el perfil de metilación de los pacientes con los hallazgos citogenéticos.
Materiales y métodosSe evaluaron 56 pacientes con leucemias: 24 con LLA, 16 con LMA y 16 con LMC. El ADN extraído se modificó con bisulfito de sodio. Se realizó un análisis de metilación en los genes CDKN2B y DBC1 mediante la PCR específica de metilación (MS-PCR). Las muestras positivas por la técnica MS-PCR fueron secuenciadas.
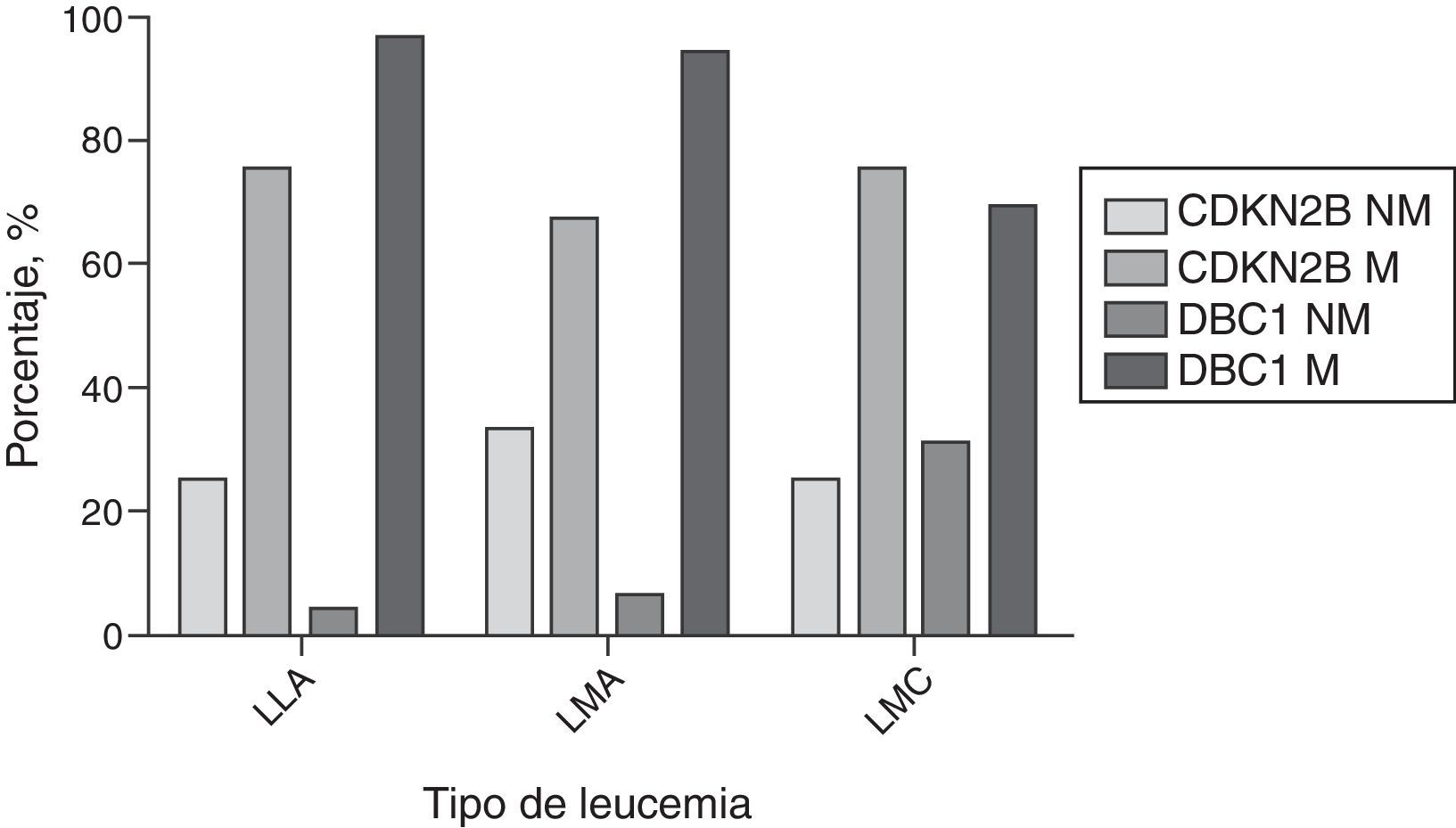
ResultadosSe encontró una frecuencia total de metilación del 87,5%. El gen CDKN2B se encontró metilado en el 75% de LLA y de LMC, y del 62% en LMA. El gen DBC1 se encontró metilado en el 96% de LLA, el 94% de LMA y del 68,8% en LMC. El gen más frecuentemente metilado en todas las muestras fue DBC1. De los tres tipos de leucemias, la LLA fue la que presentó los mayores porcentajes de metilación. El 62,5% de la muestras tenían metilado ambos genes. Las muestras con cariotipo normal presentaron una alta frecuencia de metilación de CDKN2B y DBC1.
ConclusionesEn este estudio se demostró, por primera vez en pacientes colombianos con leucemias, que la metilación de los genes CDKN2B y DBC1 es un evento frecuente. Los hallazgos indican que la metilación de genes supresores de tumores es una vía molecular alterna que podría estar relacionada con el desarrollo de neoplasias hematológicas.
To perform a methylation analysis in the CDKN2B and DBC1 gene promoters in samples from Colombian patients with acute lymphoblastic leukaemia (ALL), acute myeloid leukaemia (AML), and chronic myeloid leukaemia (CML), and to correlate the methylation profile with cytogenetic findings.
Material and methodsThe study included a total of 56 bone marrow samples, 24 from patients with ALL, 16 from AML patients, and 16 from CML patients. DNA was extracted from these samples and converted with sodium bisulphite. Methylation analysis was performed using methylation specific PCR (MS-PCR). The samples that were positive for MS-PCR were sequenced to confirm the results.
ResultsA total methylation frequency of 87.5% was found. CDKN2B gene promoter hypermethylation was found in 75% of ALL and CML samples, and 62% in AML; while DBC1 gene promoter hypermethylation was found in 96% of the samples of ALL, 94% of AML, and in 68.8% of CML. The most frequently methylated gene in all samples was DBC1. ALL was the type of leukaemia that had the highest percentages of methylation. Almost two-thirds (62.5%) of the samples had both methylated genes. Samples with normal karyotype had a high frequency of methylation in CDKN2B and DBC1 genes.
ConclusionsThis study showed, for the first time in Colombian patients with leukaemia, that methylation of DBC1 and CDKN2B genes is a common event. Our findings indicate that methylation of tumour suppressor genes is an alternate genetic pathway related to the development of haematological malignancies.
Las leucemias son enfermedades malignas que se caracterizan por la expansión clonal de las células hematopoyéticas, las cuales presentan ventajas en la proliferación, el desarrollo y la progresión de las leucemias son procesos complejos (1). Los subtipos de leucemias más comunes, de acuerdo con los tipos de células afectadas y a la progresión de la enfermedad, son: la leucemia linfoblástica aguda (LLA), la leucemia linfocítica crónica (LLC), la leucemia mieloide aguda (LMA) y la leucemia mieloide crónica (LMC)1.
Tanto factores genéticos como epigenéticos se asocian con la patogénesis de las leucemias y se reconoce que la adquisición de alteraciones genéticas en las diferentes fases tiene una gran importancia en el desarrollo de las neoplasias hematológicas2,3. Estas alteraciones comprenden: mutaciones puntuales; amplificaciones génicas; y anomalías cromosómicas estructurales como deleciones, inversiones, traslocaciones y aneuploidías, que pueden ocasionar la inactivación o la activación de diversos genes; dichas alteraciones afectan la supervivencia y la apoptosis de las células progenitoras4.
De otro lado, los cambios epigenéticos consisten en modificaciones reversibles que afectan la expresión de diversos genes sin alterar la secuencia del ADN y que puede deberse a tres fenómenos diferentes: metilación en el ADN, modificaciones en las histonas y en los ARN no codificantes (ncRNA)5. El cambio epigenético más ampliamente estudiado es la hipermetilación de los promotores de genes supresores tumorales (GST), que se asocia con la inactivación génica que ocurre por inhibición de la transcripción6.
La metilación del ADN es un fenómeno frecuente en leucemias y tumores sólidos. Se presenta principalmente en las islas CpG localizadas en la región promotora de los genes, por una modificación covalente que resulta de la actividad de una familia de enzimas denominada ADN metil-transferasas (DNMT) que catalizan la transferencia de un grupo metilo desde la S-adenosil metionina hacia el carbono 5 de los residuos de citosinas en los dinucleótidos CpG de genes relacionados con: el control del ciclo celular, la reparación del ADN, la apoptosis, la angiogénesis y la metástasis5–8. La hipermetilación desregula la expresión de diversos genes que intervienen en múltiples vías de transducción celular lo que contribuye a la carcinogénesis y a la inestabilidad cromosómica en las células progenitoras de leucemias7.
En general, la vía epigenética puede afectar diferentes mecanismos moleculares involucrados en la diferenciación, la apoptosis, la inmortalización y la transformación celular.
Estudios en leucemia informan diversos GST hipermetilados que se asocian con la transformación, el desarrollo y la progresión de esta enfermedad. Entre los GST más frecuentemente hipermetilados están: CDKN2B, CDKN1B, DBC1, RB1, entre otros9–11. Específicamente, la hipermetilación del gen CDKN2B/p15 es el más comúnmente estudiado en neoplasias hematológicas; en síndromes mielodisplásicos se ha encontrado hipermetilado este gen en el 50% de los casos.
Además, se reporta que la metilación se adquiere durante la progresión de la enfermedad y se asocia con la transformación hacia leucemia aguda, la cual es de mal pronóstico10,12,13. Por otro lado, la hipermetilación del gen DBC1, en pacientes con LMA y con cariotipo normal, se propone como un biomarcador predictivo de la supervivencia libre de enfermedad y la sobrevida global10. La hipermetilación de este gen también se informa en pacientes con LMC11. Además, se ha encontrado que la inactivación de DBC1 conduce a la desregulación del ciclo celular y a la apoptosis independiente de las caspasas11,14.
Por otra parte, la mayoría de los estudios de metilación en leucemia analizan pocos GST, sin embargo, en los últimos años, con la introducción de nuevas plataformas tecnológicas, los estudios en leucemia se han interesado en la caracterización epigenómica empleando técnicas novedosas como la de los microarrays con la que es posible evaluar el perfil de metilación de muchos genes, entre los que se incluyen genes relacionados con la iniciación y la progresión de la enfermedad, y también establecer una mejor clasificación molecular de los pacientes con malignidades hematológicas10,15–17.
Los estudios de epigenética en neoplasias hematológicas tienen un gran interés porque la hipermetilación de GST podría utilizarse como biomarcadores para el pronóstico de la enfermedad y la predicción a la respuestas de ciertos fármacos18, dado que las modificaciones epigenéticas son potencialmente reversibles y podrían convertirse en blancos moleculares para el desarrollo de nuevos agentes demetilantes para el tratamiento de pacientes con diferentes neoplasias; de esta manera, estos fármacos podrían revertir la represión génica de los GST18,19. Sin embargo, el impacto del perfil de genes hipermetilados con el pronóstico en las malignidades hematológicas aún no está bien definido.
El objetivo de este estudio es analizar la metilación en los promotores de los genes DBC1 y CDKN2B en muestras de pacientes colombianos con diagnóstico de LLA, LMA y LMC y correlacionar el perfil de metilación de los pacientes con los hallazgos citogenéticos.
MétodosPacientes y muestrasSe realizó un estudio descriptivo de tipo prospectivo. La población de estudio estuvo constituida por 56 pacientes con diagnóstico clínico y hematológico de leucemia: 16 con LMA, 16 con LMC y 24 con LLA. De los cuales, 49 pacientes tenían diagnósticos de novo, 4 presentaban recaídas y 3 estaban en tratamiento; 25 de los pacientes eran mujeres y 31 eran hombres.
Las muestras de medula ósea o de sangre periférica de los pacientes fueron obtenidas por los hematólogos en diferentes instituciones de salud y remitidas al laboratorio de Genética Médica de la Facultad de Medicina de la Universidad de Antioquia, durante 2011 y 2012. Los pacientes firmaron el consentimiento aprobado por el Comité de Bioética, Sede de Investigación Universitaria de la Universidad de Antioquia, de la ciudad de Medellín. Adicionalmente, se incluyeron 18 muestras de sangre periférica de individuos sanos para control.
Extracción de ADNLa extracción del ADN genómico se realizó a partir del aspirado de medula ósea o de sangre periférica de los pacientes y de los individuos sanos, utilizando el kit comercial QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) y siguiendo las recomendaciones de la casa comercial.
El ADN de cada muestra se cuantificó en un espectrofotómetro NanoDrop 2000c Spectrophotometer (Thermo Scientific, USA). La calidad y la integridad del ADN se verificó en geles de agarosa al 2% teñidos con bromuro de Etidio. Las bandas se visualizaron con luz UV en un fotodocumentador de geles. El DNA extraído se almacenó a -20°C.
Conversión del ADN con bisulfito de sodioEl DNA extraído se modificó con bisulfito de sodio, usando el kit comercial EpiTect Bisulfite Kits (Qiagen, Hilden, Germany), de acuerdo con las instrucciones del fabricante. El ADN modificado se resuspendió en agua y se almacenó a–20°C hasta su uso. El tratamiento con bisulfito de sodio modificó las citosinas no metiladas y que son convertidas en uracilos, mientras que las citocinas metiladas no fueron modificadas.
Amplificación de ADN por PCR específica de metilación (MS-PCR)La técnica de MS-PCR se utilizó para determinar la metilación en los promotores de los genes CDKN2B y DBC1. El ADN modificado con bisulfito de sodio se amplificó por PCR en un termociclador Gene Amp PCR System 9700 (Applied Biosystems, USA). La MS-PCR para los dos genes se realizó en un volumen total de 25μl que contenía 200ng de ADN modificado. Las secuencias de los cebadores utilizados para la amplificación de los promotores de cada gen y las condiciones de la MS-PCR se realizaron siguiendo protocolos previamente descritos10; para cada gen se utilizó un par de cebadores que reconocen en el promotor regiones metiladas y otro par para regiones no metiladas. En la MS-PCR se utilizó como control negativo DNA a partir de linfocitos de individuos sanos y como control positivo se utilizó un DNA genómico universalmente metilado in vitro con la enzima ADN metil transferasa (New England, Biolabs). Los productos amplificados por la MS-PCR se corrieron en una electroforesis en geles de agarosa al 2%, teñidos con bromuro de Etidio. Las bandas se visualizaron directamente en un fotodocumentador de geles.
SecuenciamientoLos productos amplificados se purificaron con el Kit Wizard DNA purification (Promega) y secuenciaron directamente por ambas cadenas en un analizador genético automático 3730xl DNA Analyzer (Applied Biosystems).
Análisis de resultadosLas secuencias obtenidas se editaron y analizaron con los programas ContigExpress, Vector NTI y AlignX. En el ContigExpress se ensamblaron las secuencias obtenidas con los cebadores ¿forward¿ y ¿reverse¿ para formar un contig de cada muestra. Una vez obtenido el contig, se utilizó el programa Vector NTI para realizar la edición de las secuencias y finalmente, en el programa AlignX, se alineó la secuencia editada de cada muestra con la secuencia de referencia para cada gen que se encuentran en el Genbank (National Center for Biotechnology Information). Para el gen DBC1 se usó la secuencia NM_014618 y para CDKN2B la secuencia NM_078487.2.
Análisis estadísticoLos resultados se analizaron mediante el programa SPSS versión 19. En la estadística descriptiva para variables cuantitativas se utilizó la media y el rango; las variables categóricas se presentaron en frecuencias y porcentajes. Los resultados de la MS-PCR se analizaron como una variable dicotómica, con base en la presencia o la ausencia de metilación del gen. Los resultados de las muestras de leucemia y de los controles se compararon mediante la prueba exacta de Fischer. Se utilizó el valor de p bilateral. Se consideró significativo un valor de p< 0,05.
ResultadosSe estudió un total de 56 pacientes con diagnóstico de leucemia: 24 (42,9%) con LLA, 16 (28,6%) con LMA y 16 (28,6%) con LMC. Con relación al género, el 55,4% (31/56) de los pacientes eran hombres y el 44,6% (25/56) eran mujeres. La edad promedio para ambos sexos fue de 36 años.
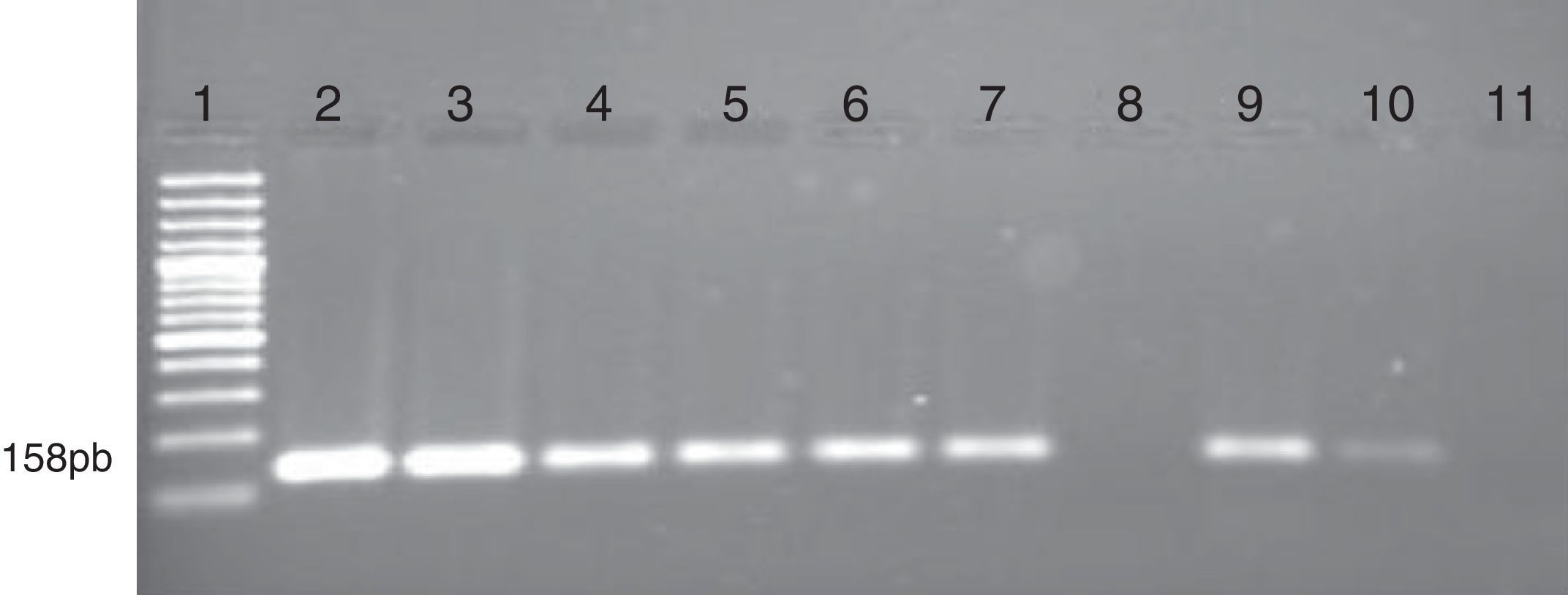
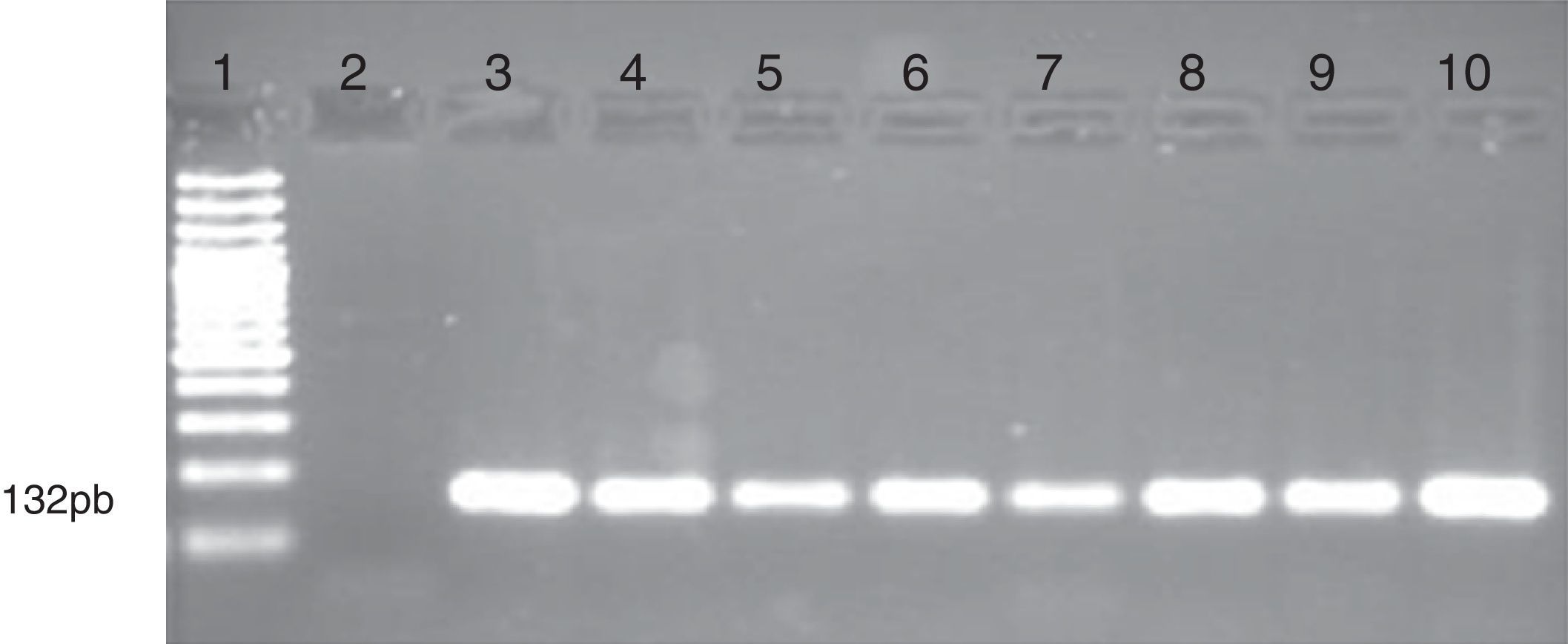
Metilación de los genes CDKN2B y DBC1 en pacientes con leucemiasLa determinación de la metilación de los promotores de los genes CDKN2B y DBC1 en los pacientes con leucemia analizados se realizó mediante las amplificaciones de regiones metiladas y no metiladas por la técnica MS-PCR. En las figuras 1 y 2 se muestran los resultados del estado de metilación de estos dos genes en algunas muestras de pacientes con leucemia.
. Carril 2 control positivo (ADN metilado universalmente; Epitec control DNA, QIAGEN). Carriles 3 al 7, 9 y 10 corresponde a pacientes con metilación del gen DBC1. Carril 8 paciente que no presenta metilación y carril 11 control negativo (sin DNA).")
Electroforesis en gel de agarosa que muestra el análisis de metilación del gen DBC1 por MS-PCR en muestras de pacientes con leucemias. Se observa las bandas obtenidas con los cebadores específicos que amplifican las regiones metiladas del promotor del gen. Carril 1 marcador de peso molecular de 100pb (GeneRuler 100bp DNA Ladder, Fermentas). Carril 2 control positivo (ADN metilado universalmente; Epitec control DNA, QIAGEN). Carriles 3 al 7, 9 y 10 corresponde a pacientes con metilación del gen DBC1. Carril 8 paciente que no presenta metilación y carril 11 control negativo (sin DNA).
. Carril 2 control negativo (sin DNA). Carril 3 control positivo (ADN metilado universalmente; Epitec control DNA, QIAGEN). Carriles 4 al 10 corresponde a pacientes con metilación del gen CDKN2B.")
Electroforesis en gel de agarosa que muestra el análisis de metilación del gen CDKN2B por MS-PCR en muestras de pacientes con leucemias. Se observa las bandas obtenidas con los cebadores específicos que amplifican las regiones metiladas del promotor del gen. Carril 1 marcador de peso molecular de 100pb (GeneRuler 100bp DNA Ladder, Fermentas). Carril 2 control negativo (sin DNA). Carril 3 control positivo (ADN metilado universalmente; Epitec control DNA, QIAGEN). Carriles 4 al 10 corresponde a pacientes con metilación del gen CDKN2B.
La frecuencia total de metilación en las 56 muestras de leucemias analizadas fue del 87,5% (49/56) y el 12,5% (7/56) de las muestras no presentó metilación. Todos los siete casos de los pacientes que al momento del estudio estaban en recaída o tratamiento presentaron metilación en los genes evaluados, similar a los resultados obtenidos en los pacientes con diagnóstico de novo. La frecuencia de metilación del gen CDKN2B fue del 71,4% (40/56) y para el gen DBC1 fue del 87,5% (49/56). En general, comparando los resultados de metilación de los dos genes con todos los casos de leucemia analizados, se encontró que el gen DBC1 presentó la mayor frecuencia de metilación con una diferencia estadísticamente significativa (p=0,035). En las muestras de los controles no se detectó metilación en estos dos genes.
Metilación en pacientes con LLAEn este tipo de leucemia el 37% (9/24) eran hombres y el 63% (15/24) mujeres; 37% (9/24) eran niños; la edad promedio fue de 25,7 años (rango 3-84). La frecuencia de metilación del gen CDKN2B en estos pacientes fue del 75% (18/24), mientras que para el gen DBC1 la frecuencia de metilación fue del 96% (23/24) (fig. 3) y solo en una muestra (4%) no se detectó metilación. Se encontró que el 29% (7/24) de las muestras tenían metilación en el promotor de un solo gen, mientras que el 71% (17/24) presentó metilación en los dos genes y no se encontraron diferencias significativas (p=0,14). En todas las muestras de los niños (9/9) se presentó metilación y con una mayor frecuencia en el gen DBC1 (6/9). No se encontraron diferencias significativas entre los resultados de metilación de niños y adultos (p=1,0).
Metilación en pacientes con LMA
En este grupo el 75% (12/16) eran hombres y el 25% (4/16) mujeres; el 12,5% (2/16) eran niños; la edad promedio fue de 36,5 años (rango 2-70). La frecuencia de metilación del gen CDKN2B fue del 62% (10/16), mientras que la frecuencia de metilación del gen DBC1 fue del 94% (15/16) (fig. 3); una sola muestra (6%) no estaba metilada. El 44% (7/16) de las muestras analizadas presentaron metilación en un solo gen y el 56% (9/16) tenía metilado ambos genes y no se encontraron diferencias significativas (p=0,08). Los dos casos de niños estaban metilados.
Metilación en pacientes con LMCEn este grupo se encontró que el 75% (12/16) eran hombres y el 25% (4/16) eran mujeres; la edad promedio fue de 52 años (rango 18-79). La frecuencia de metilación del gen CDKN2B fue del 75% (12/16) y del gen DBC1 fue del 68,8% (11/16) (fig. 3). De otro lado, se encontró que el 31,2% (5/16) de las muestras analizadas presentaron metilación en el promotor de un solo gen y el 56,3% (9/16) mostró metilación en ambos genes y no se encontraron diferencias significativas (p=0,70). Además, se encontró que el 12,5% (2/16) de las muestras analizadas no tenía metilación en ninguno de los dos genes evaluados.
Cariotipo de los pacientes con leucemiasEl 48,2% (27/56) de los pacientes tenían cariotipo normal y el 41,1% (23/56) presentó cariotipo anormal; en el 10,7% (6/56) de las muestras no se obtuvo el resultado del cariotipo. En las muestras de LLA con cariotipo normal se encontró una frecuencia de metilación de los genes CDKN2B y DBC1 del 69% (11/16) y 100% (16/16) respectivamente, y no se encontraron diferencias (p=1,0). En las muestras de LMA fue del 78% (7/9) y 89% (8/9) para CDKN2B y DBC1 respectivamente, y no se encontraron diferencias (p=0,22).
En las muestras de LMC con cariotipo normal la metilación de estos dos genes fue un evento infrecuente; no obstante, en este tipo de leucemia se presentó la mayor frecuencia de cariotipos anormales, seguido del grupo con LMA. Por último, en el grupo de pacientes con cariotipo anormal se encontró que la alteración cromosómica más común fue la de tipo estructural (translocación).
Para confirmar los resultados de la metilación obtenidos por la MS-PCR, se secuenciaron 20 muestras de cada gen seleccionadas al azar. En los cromatogramas analizados de la región promotora, que contienen las islas de dinucleótidos CpG, se confirmó la metilación de las islas CpG en todos los casos. Asimismo, mediante el secuenciamiento directo de las muestras no metiladas se comprobó la conversión completa con bisulfito de sodio de las C no metiladas en T en las islas CpG.
En los análisis del secuenciamiento de los promotores para determinar la metilación se encontró un polimorfismo en el gen DBC1, que está reportado en el Genbank con el código rs2065399. Este polimorfismo se identificó en las muestras de LLA, LMA y LMC.
En resumen, los resultados de este trabajo mostraron una alta frecuencia de metilación de los genes CDKN2B y DBC1 en los pacientes con LLA, LMA y LMC estudiados; el gen más frecuentemente metilado fue el DBC1 (p=0,035). Adicionalmente, se encontró que la metilación de los genes DBC1 y CDKN2B es más frecuente en pacientes con LLA y LMA, que tenían cariotipo normal.
DiscusiónEste es el primer estudio de análisis de metilación en los promotores de los genes DBC1 y CDKN2B por MS-PCR que se realiza en 56 pacientes con LLA, LMA y LMC en Latinoamérica. En total, se encontró un alto porcentaje de metilación (87,5%) en las muestras analizadas.
En este trabajo se demostró una alta frecuencia de metilación de los genes DBC1 y CDKN2B en los diferentes tipos de leucemia evaluados. Estos resultados concuerdan con los informados por otros estudios, en los que reportan un alto porcentaje de metilación de estos genes en neoplasias hematológicas11,12,14,15,20,21. Por lo tanto, el perfil de metilación de los casos con recaída y tratamiento fueron similares a los obtenidos con los casos de novo y con los resultados obtenidos se confirmó que la metilación de los promotores de los genes DBC1 y CDKN2B es un evento común en la patogénesis de la LLA, la LMA y la LMC.
En las muestras de LLA se encontró que la metilación de los genes DBC1 y CDKN2B fue muy frecuente, con mayor predominio en el primer gen y no se encontraron diferencias significativas. Estos resultados son consistentes con los reportados por Grønbæk et al., quienes obtuvieron una frecuencia de metilación del 100% en el gen DBC114. Por el contrario, en otro estudio que incluyó 170 muestras de pacientes con LLA11, se encontró una baja frecuencia de metilación para este gen comparada con la reportada en este estudio. No obstante, en este mismo trabajo, se encontró una alta frecuencia de metilación de DBC1 en 4 líneas celulares de LLA, similar a lo informado en este trabajo. Las diferencias encontradas en los anteriores resultados podrían explicarse por el tamaño de la muestra, el cual fue menor al usado y a diferencias poblacionales en las frecuencias reportadas para este gen. De otro lado, se informa que la hipermetilación del gen DBC1 (The Deleted in Bladder Cancer 1 gene), localizado en la región cromosómica 9q33.1, es muy frecuente en cáncer de vejiga, pulmón y cavidad oral22–24. La hipermetilación no permite la expresión de este gen en las células tumorales, por lo que se sugiere que la metilación de DBC1 tiene una gran importancia en el desarrollo de LLA, de algunos tumores sólidos10,11,22,23 y se asocia con un mal pronóstico de la enfermedad25.
De otro lado, estudios realizados en metilación del gen CDKN2B en muestras de LLA informan un rango de frecuencia de metilación del 34% hasta el 100%21,26,27. La frecuencia obtenida en este trabajo está dentro de este rango y concuerda con lo informado en la literatura. Dado que los anteriores resultados se han obtenido de pacientes con LLA de diferentes poblaciones, se sugiere que las diferencias en la frecuencia de metilación del gen CDKN2B reportadas podrían deberse, además del tamaño de la muestra, a variaciones geográficas. El gen CDKN2B es uno de los genes más ampliamente estudiados en los mecanismos epigenéticos en neoplasias hematológicas y es el que se ha encontrado más frecuentemente hipermetilado12,20,27.
En el estudio efectuado se encontró un alto porcentaje de metilación en el promotor del gen CDKN2B en pacientes colombianos con la LLA, por lo que estos hallazgos sugieren que este gen podría estar involucrado en la patogénesis de la LLA.
En cuanto a los resultados de metilación en las muestras de LMA, se encontró, similar a lo obtenido en la LLA, una alta frecuencia de metilación del 94% en DBC1, mayor al 52% informado por Álvarez et al., en 244 muestras analizadas10.
Hasta la fecha, el estudio de estos autores es el único que se encuentra en la literatura sobre metilación del gen DBC1 en pacientes con LMA; lo que indica que nuestro trabajo es el primero en reportar el mayor porcentaje de metilación para este gen y el segundo en informar el estado de metilación del promotor del gen DBC1 en LMA.
En el trabajo de Álvarez et al.10, en un subgrupo de pacientes con LMA se encontró que tenían cariotipo normal y una asociación de la hipermetilación del gen DBC1 con una menor tasa de supervivencia libre de la enfermedad comparado con un grupo de pacientes que no tenían metilado este gen. A partir de estos hallazgos concluyeron que la hipermetilación del gen DBC1, en este grupo de pacientes, podría considerarse como un marcador pronóstico de gran utilidad clínica para esta enfermedad.
En el estudio realizado se encontró en el grupo de pacientes con LMA y cariotipo normal una alta frecuencia de metilación similar a la reportada por Álvarez et al10, pero no se encontraron diferencias estadísticas significativas para esta asociación, posiblemente debido al tamaño de la muestra. Debe tenerse en cuenta que en este, además, de ser el primero en Colombia, no fue posible establecer la sobrevida de la enfermedad en los pacientes evaluados por lo que se requiere de futuros estudios que validen los hallazgos presentados.
A diferencia de la poca información que existe de la metilación de DBC1 en LMA, para el gen CDKN2B se tiene más conocimientos de la metilación en este tipo de leucemia. Las frecuencias de metilación informadas por varios estudios para CDKN2B están en el rango de 40 al 93%10,20,28–30. En este estudio se encontró una frecuencia del 62%, este resultado concuerda con el rango informado. No obstante, la frecuencia reportada en este trabajo es mayor a la informada en otros estudios en los que analizaron un mayor número de muestras10,29,30; tampoco no se encontró asociación estadísticamente significativa en los pacientes con LMA respecto al estado de metilación del gen CDKN2B.
Cabe mencionar que en la población latinoamericana solo se han realizado dos trabajos de metilación en LMA. Los resultados presentados concuerdan con los reportados por Santos et al. en Brasil30, pero difieren de los de Reyes et al.10 en Chile, quienes informaron una frecuencia de metilación para CDKN2B tres veces menor a la reportada en este estudio con pacientes colombianos. De los anteriores resultados se podría concluir que se presenta una variación geográfica en la frecuencia de metilación de CDKN2B en pacientes con LMA.
En resumen, los diferentes estudios de metilación en este tipo de leucemia concluyen que el gen CDKN2B en la mayoría de los pacientes con LMA se encuentra hipermetilado y que este mecanismo epigenético se podría asociar como un mayor riesgo para el desarrollo de este tipo de leucemia10,30; para corroborar la anterior asociación en los pacientes colombianos con LMA es necesario realizar más estudios que incluyan un mayor número de pacientes y en los que se determine la tasa de sobrevida de los pacientes.
En cuanto a los hallazgos de metilación en las muestras de LMC, en este trabajo, se demostró nuevamente una alta frecuencia de metilación en los dos genes estudiados, similar a lo observado en LLA y LMA. En la literatura existen pocos estudios sobre la metilación en pacientes con LMC, lo cual podría explicarse en parte porque la translocación t (9;22), que corresponde a la alteración cromosómica recurrente que se presenta en estos pacientes y se relaciona directamente con el origen de la LMC2,32. La detección de este marcador cromosómico usualmente se realiza por métodos de citogenética convencional, FISH o qPCR2,18. Con respecto a la metilación del gen DBC1 en LMC, solo se encuentra publicado un estudio con cinco pacientes, que reporta metilación en todos los casos15. Este resultado concuerda con el obtenido en el estudio presentado y que incluyó un mayor número de muestras; es el segundo estudio en informar que en pacientes con LMC se presenta un porcentaje alto de metilación DBC1. De estos hallazgos podría sugerirse que este mecanismo epigenético ocurre de forma independiente al de la alteración cromósomica t (9;22) y que también podría estar relacionado con el desarrollo de la LMC. Sin embargo, se requiere de más estudios para validar estos resultados.
Contrario a lo informado para el gen DBC1 en LMC, varios estudios reportan bajas frecuencias de metilación del gen CDKN2B, desde 0 al 24%15,33–36. Estos resultados no concuerdan con lo observado en el presente trabajo, en el que se obtuvo una alta frecuencia de metilación para este gen. A diferencia de lo que se informa en LMC para el gen CDKN2B, este se encuentra metilado con mayor frecuencia en LLA, LMA y SMD12,13,20,21; los autores concluyen que en la LMC la metilación de CDKN2B es un evento infrecuente. De todo lo anterior, podría afirmarse que el presente estudio sería el primero en reportar una alta frecuencia de metilación del gen CDKN2B en pacientes con LMC. No obstante, son necesarios más estudios para corroborar este hallazgo.
Las diferencias encontradas en los resultados podrían deberse, además, del tamaño de la muestra, a la metodología empleada en cada estudio y al tipo de población estudiada. En conclusión, similar a lo encontrado en la LLA y la LMA, en la LMC también se demostró una alta frecuencia de metilación de los genes DBC1 y CDKN2B.
Adicionalmente, los estudios de metilación de CDKN2B en LMC, también han evaluado el perfil de metilación en las diferentes fases de la enfermedad. Nguyen et al.33, reportaron en la fase crónica un 7% de metilación, en la fase acelerada un 9% de metilación y en crisis blástica un 16% de metilación. Por otro lado, Jelinek et al.18 informan en la fase crónica un 2% de metilación, en fase acelerada un 17% de metilación y en crisis blástica un 27% de metilación.
En el estudio realizado todos los pacientes con LMC se encontraban en crisis blástica lo que podría explicar, en parte, el alto porcentaje de metilación encontrado en los genes DBC1 y CDKN2B. En resumen, se sugiere que el estado de metilación del gen CDKN2B aumenta con la progresión de la enfermedad. Son necesarios más estudios para establecer esta relación. De otro lado, Jelinek et al18 también proponen que en los pacientes con LMC la inactivación de genes supresores de tumores, como CDKN2B por mecanismos epigenéticos, podría ser una de las causas de resistencia al Imatinib, un medicamento antineoplásico de primera línea utilizado en el tratamiento de esta enfermedad.
Por otra parte, en cuanto a la relación de la metilación de los genes DBC1 y CDKN2B con el cariotipo, se observó que la mayoría de los pacientes con cariotipo normal tenían metilados ambos genes, similar a lo reportado por otros autores10,20,21,29,37,38. Sin embargo, en este estudio no se encontraron diferencias significativas para dicha asociación, posiblemente por el tamaño de la muestra. Se menciona que otros estudios no logran establecer esta relación11,39. Se destaca que DBC1 fue el gen más comúnmente metilado en los tres tipos de leucemias analizadas, este fue el único resultado del presente estudio que mostró una diferencia estadísticamente significativa (p=0,035). Este hallazgo es importante porque permitiría en futuros estudios proponer al gen DBC1 como un biomarcador epigenético dentro de la patogenésis de las leucemias en pacientes colombianos y su relación con el pronóstico de la enfermedad. Por otra parte, también se ha informado que la inestabilidad cromosómica y las modificaciones epigenéticas presentan una correlación negativa40, tal como se observó en este estudio con el grupo de pacientes con LMC. De estos hallazgos se resalta la importancia de estudiar el efecto de la metilación en malignidades hematológicas como una vía diferente relacionada con el desarrollo de estas enfermedades, especialmente en los casos que presentan cariotipo normal porque sería muy útil para el diagnóstico y el pronóstico de las enfermedades17,41.
La metilación de novo de las islas CpG localizadas en los promotores de los genes DBC1 y CDKN2B es un mecanismo epigenético frecuente que ocurre en neoplasias hematológicas y tumores sólidos, cuyo resultado es la inactivación génica3,6,13,14,11,22. Estos dos genes supresores de tumores tienen una regulación negativa del ciclo celular por lo que la inactivación se asocia con una ventaja en el crecimiento, la proliferación celular y en la trasformación maligna42,43. Además, la metilación de las islas CpG podría ocurrir en etapas tempranas del desarrollo de las neoplasias hematológicas. Por lo anteriormente mencionado, varios estudios reportan que la metilación de los genes DBC1 y CDKN2B se relaciona con un mayor riesgo de desarrollar malignidades hematológicas8,10,11,13,14,17,31,40,41; no obstante, la utilidad clínica del perfil de metilación en diversos genes es controvertida, por lo que se requiere de futuros estudios para demostrar esta relación.
En el estudio realizado se encontró que la mayoría de las muestras analizadas presentaban metilación en estos dos genes, por lo que se sugiere que la inactivación de ambos genes podría estar relacionada con la patogénesis de LLA, LMA y LMC en estos pacientes. Además, en estas mismas muestras no se podría descartar que otros genes pudieran estar metilados, puesto que se ha demostrado por estudios de metilomas con técnicas avanzadas de microarrays, que es común la metilación de múltiples genes en diversas neoplasias, los cuales se relacionan con la iniciación y la progresión del cáncer10,15–17,11,44.
En general, los estudios de metilación en cáncer son muy interesantes porque la hipermetilación de genes supresores de tumores se asocia con un mecanismo epigenético reversible y la re-expresión de los genes metilados en algunos casos en particular presentan un mejor pronóstico de la enfermedad5,6,14,19,11,23. Por esta razón, la identificación de genes hipermetilados en diferentes enfermedades genéticas tienen el potencial de ser utilizados como biomarcadores para el pronóstico de la enfermedad y también como blancos moleculares para el desarrollo de nuevos medicamentos demetilantes5,8,16,45.
Dado estos avances, a los pacientes con determinadas malignidades hematológicas se les suministran terapias epigenéticas con agentes hipometilantes, como la 5 azacitidina con la que se ha logrado obtener una mejor respuesta al tratamiento y también una mejor sobrevida7,8,19,45,46. Además, estos avances son importantes para establecer el perfil de metilación con que se podría lograr una clasificación molecular más precisa de los pacientes que se benefician de este tipo de tratamientos16,17,38,44; sin embargo, la eficacia de los agentes hipometilantes en cáncer y la utilidad clínica aún no están bien dilucidados.
Este es el primer estudio de análisis de metilación en los genes DBC1 y CDKN2B realizado en pacientes colombianos con LLA, LMA y LMC; también es el primero en Latinoamérica que evalúa el estado de metilación del gen DBC1 en leucemias. Los resultados aportan información básica importante del perfil de metilación de estos dos genes en neoplasias hematológicas para nuestra población, especialmente la alta frecuencia de metilación del gen DBC1 encontrada en las muestras de LLA, LMA y LMC. Los resultados de este estudio confirman hallazgos previos de las frecuencias de metilación de DBC1 y CDKN2B en LLA y LMA, pero difieren con las encontradas en LMC, particularmente con el gen CDKN2B, siendo el primer estudio en informar una alta frecuencia de metilación.
Las diferencias de los resultados obtenidos frente a los de otros estudios podrían explicarse por varias razones como: el tamaño de la muestra; las metodologías empleadas; la exposición a determinados agentes medioambientales y a la composición genética de la población colombiana, la cual es considerada históricamente como un aislado genético compuesto por una mezcla étnica de europeos, africanos y amerindios47.
Son necesarios futuros estudios con un mayor número de muestras y emplear técnicas epigenómicas para evaluar múltiples genes con el fin de validar estos hallazgos en la población de Colombia, como también para esclarecer el valor del diagnóstico y del pronóstico de la metilación de los genes DBC1 y CDKN2B con el riego de desarrollar malignidades hematológicas. Respecto a la carcinogénesis de las neoplasias hematológicas la vía epigenética debe tenerse en cuenta como una vía molecular alterna que podría estar relacionada con el desarrollo de estas enfermedades.
ConclusiónEn este estudio se encontraron altas frecuencias de metilación de los genes DBC1 y CDKN2B en pacientes colombianos con LLA, LMA y LMC, y especialmente las del gen DBC1 fueron mayores. Los resultados indican que la metilación de estos dos genes supresores de tumores es un evento común en estos pacientes, por lo que la vía epigenética constituye una vía molecular alterna que podría estar relacionada con el desarrollo de neoplasias hematológicas.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónEste trabajo de investigación fue financiado por la Universidad de Antioquia, Medellín, Colombia. Programa de Sostenibilidad de Grupos 2009-2010, Genética Médica, CPT- 0905.
Conflicto de interesesLos autores no tienen conflicto de interés en relación con el artículo que se remite para publicación.
A los pacientes por su participación voluntaria en este trabajo; al Hospital Universitario San Vicente Fundación de la ciudad de Medellín, Colombia, por su colaboración con la remisión de muestras. Al laboratorio de Genética Médica de la Facultad de Medicina, de la Universidad de Antioquia, por suministrar los resultados de los cariotipos de los pacientes. Al Dr. Jorge Botero Garcés, por su permanente asesoría en los análisis estadísticos.
