





Los productos naturales han sido la fuente más representativa en la obtención de agentes terapéuticos para el tratamiento del cáncer y su enorme contribución se reconoce en el descubrimiento de nuevas moléculas citotóxicas con variados mecanismos de acción. La mayoría de medicamentos utilizados para tratar el cáncer son poco selectivos y presentan altos niveles de resistencia y toxicidad, afectando considerablemente el pronóstico de vida en pacientes con esta enfermedad. Este artículo revisa los principios activos obtenidos de fuentes vegetales para el tratamiento del cáncer y sus distintos mecanismos de acción, abordando los avances más recientes en dianas terapéuticas como las proteínas inhibidoras de apoptosis (IAP) y algunas moléculas naturales estructuralmente sencillas que se encuentran en diferentes fases de estudio clínico y que son interesantes a nivel farmacológico debido a su alta selectividad, baja toxicidad y gran potencial terapéutico frente a distintos tipos de cáncer.
Natural products have been the most representative source of small molecules for cancer therapy, and have made enormous contributions to the discovery of new drugs with varied mechanisms of action. Most of these drugs are highly toxic and show low specificity to cancer cells, considerably affecting the survival prognosis in patients with this disease. The purpose of the current article is to discuss the mechanism of action of bioactive phytochemicals against cancer. The most promising therapeutic targets are presented such as the inhibitor of apoptosis proteins (IAP), as well as some bioactive small molecules of pharmacological interest and high specificity against cancer cells.
A pesar de que en los últimos años los científicos han retomado las investigaciones en productos naturales como fuente de nuevos fármacos para el tratamiento de enfermedades crónicas como el alzheimer, la artritis, la hepatitis, la diabetes, la tuberculosis, el sida y el cáncer; hoy en día muchos países en desarrollo siguen utilizando la medicina tradicional debido al bajo costo y acceso limitado a los tratamientos farmacéuticos. Estos conocimientos etnobotánicos son los responsables en muchas ocasiones del descubrimiento de nuevos principios activos frente a distintas enfermedades, por ejemplo, los compuestos naturales vinblastina, taxol, etopósido e irinotecan, entre muchos otros, han traído una nueva luz para el tratamiento de los enfermos de cáncer1. Sin embargo, la complejidad química y estructural característica de estas moléculas hacen difíciles y costosos sus procesos de aislamiento, elucidación estructural y generación de nuevas rutas sintéticas que permitan obtenerlos en mayor cantidad.
No podemos considerar como una sorpresa o simplemente como una coincidencia, el hecho de que la investigación fitoquímica represente un legado histórico importante en el descubrimiento de nuevos fármacos contra el cáncer. Los mecanismos de defensa de las plantas a menudo se basan en su capacidad de generar sustancias químicas que posean un efecto citotóxico o citostático y sus características únicas tales como la selección evolutiva de poseer actividad biológica las convierten en una fuente inestimable de nuevas sustancias con actividad antitumoral. Es así como la lista de medicamentos contra el cáncer que han proporcionado los productos naturales incluye a fármacos de gran éxito como la doxorrubicina, paclitaxel, vinblastina, etopósido, irinotecan, gemcitabina, metotrexato2; y fitofármacos como el polifenon E, que es una mezcla estandarizada de los polifenoles epicatequina (EC), epigalocatequina (EGC), galato de epicatequina (ECG), galato de epigalocatequina (EGCG) y galato de galocatequina (GCG), preparada de las hojas del árbol de té Camellia sinensis (Theaceae)3. Muchos de estos fármacos son utilizados actualmente para el tratamiento de diferentes tipos de cáncer; sin embargo, aún quedan problemas por superar como son los altos costos, la resistencia y la toxicidad de algunos medicamentos. Según la Organización Mundial de la Salud, en el 2012 se reportaron 14,1 millones de casos nuevos de cáncer, 8,2 millones de muertes y se estima que para el 2030 habrá 26,4 millones de personas con esta enfermedad4.
Numerosas investigaciones han reiterado y confirmado la importancia que tienen los productos de origen vegetal en la búsqueda de moléculas de interés terapéutico frente al cáncer5–10. La primera generación de medicamentos descubiertos contra el cáncer se centraba en la obtención de moléculas que actuarán como agentes citotóxicos, bien sea que causarán daños irreparables al ADN, inhibiendo su síntesis o interfiriendo con los mecanismos de división celular; por ejemplo, bloqueando las topoisomerasas o la unión de microtúbulos11. Muchos de estos agentes fueron descubiertos realizando ensayos biológicos basados únicamente en compuestos que fueran capaces de atacar células tumorales, encontrando en los productos naturales vegetales una fuente de moléculas activas con una gran variedad de núcleos estructurales como alcaloides, lignanos, flavonoides, estilbenos, taxoides, combrestatinas, acetogeninas y quinonas. Esta revisión presenta un panorama general en el desarrollo de fármacos para tratar el cáncer basados en productos naturales vegetales, diferenciando los mecanismos de acción poco selectivos a nivel celular de algunos de los fármacos tradicionalmente más usados y los mecanismos de acción que pueden ser selectivos a líneas celulares tumorales, a través de avances específicos en dianas terapéuticas como las proteínas inhibidoras de apoptosis (IAP), en especial la proteína XIAP (por sus siglas en inglés, X-linked Inhibitor of Apoptosis), o el aumento de especies reactivas de oxígeno en la célula que involucran la inhibición de enzimas de respuesta al estrés oxidativo como la glutatión s-transferasa (GSTP1) y la carbonil reductasa (CBR1). Se revisaron libros y artículos científicos que fueron publicados entre los años 2000 y 2015 en las bases de datos ScienceDirect, PubMed, American Chemical Society y Nature, con las palabras: anticancer, antitumoral, antineoplásico, actividad citotóxica, producto natural, etnobotánica, antagonistas de IAP y antagonista de XIAP.
Compuestos vegetales utilizados en el tratamiento del cáncerLa importancia de los productos naturales en el desarrollo de nuevos fármacos, radica en que el 75% de los medicamentos utilizados para tratar el cáncer y algunas enfermedades infecciosas durante los años de 1981 a 2002, fueron derivados de productos naturales; y más adelante entre el 2002 y el 2005, gracias a las fuentes naturales fueron introducidos al mercado 23 nuevas medicinas para el tratamiento de diferentes enfermedades12. En los años 2007 y 2008 salieron al mercado 55 nuevos medicamentos derivados de productos naturales, de los cuales, 16 fueron aislados de fuentes naturales y 39 derivados de estructuras químicas obtenidas de ellas13. De hecho, si de los 206 medicamentos aprobados para tratar el cáncer entre los años de 1940 y 2010 se excluyen los que tienen principios activos de alto peso molecular, quedarían 175 medicamentos, de los cuales 131 (aproximadamente el 75%) tienen que ver con productos naturales, bien sea porque se obtienen directamente de una fuente natural, o se obtienen por síntesis química de una estructura natural, o se hacen pequeñas modificaciones químicas a compuestos obtenidos de una fuente natural14.
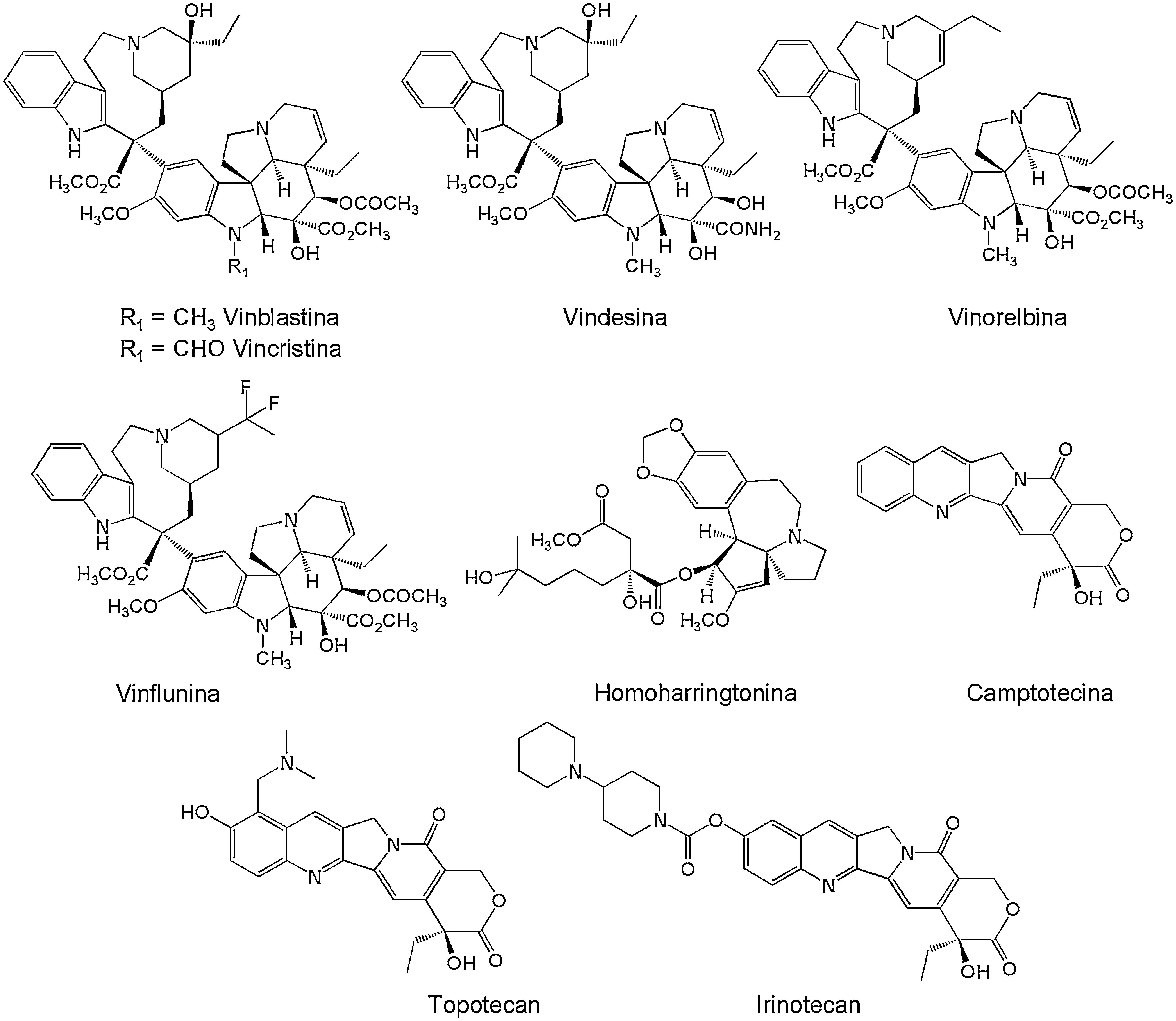
Dentro de los compuestos naturales vegetales más utilizados para el tratamiento del cáncer de mama, testículo, pulmón y leucemia, se encuentran diferentes alcaloides (fig. 1) como la vinblastina y vincristina, aislados de la especie vegetal Catharanthus roseus (Apocynaceae) endémica de Madagascar15; la camptotecina, aislada de la especie Camptotheca acuminata (Cornaceae)16; y la homoharringtonina, aislada del árbol de hoja perenne Cephalotaxus harringtonia K. Koch var. Harringtonia (Cephalotaxaceae) originario de Japón. A partir de estos compuestos se han desarrollado derivados semisintéticos que mejoran su actividad o solubilidad como lo son la vindesina, vinorelbina, vinflunina, topotecan e irinotecan (fig. 1), siendo estos dos últimos comercializados como Hycamtin y Campostar por GlaxoSmithKline y Pfizer, para los tratamientos de cáncer de colon y ovario5,17.
 y análogos sintéticos (vindesina, vinolerbina, vinflunina, topotecan, irinotecan).")
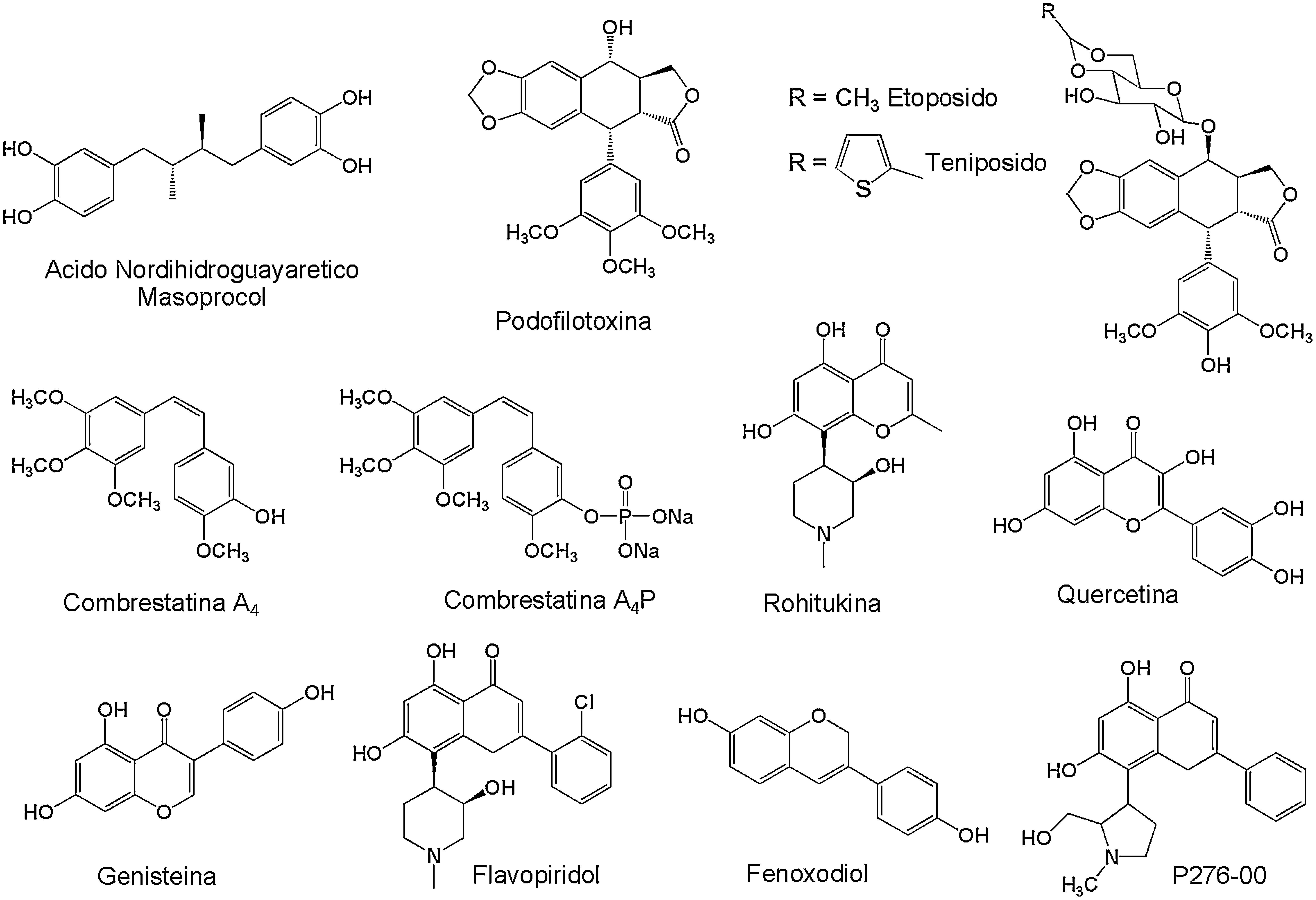
Dentro de los compuestos de tipo lignano (fig. 2), se encuentran el ácido nordihidroguayaretico aislado de la especie Larrea tridentata (Zygophyllaceae) y la podofilotoxina aislada de especies vegetales del género Podophyllum (Berberidaceae), principalmente Podophyllum peltatum y Podophyllum notatum, a partir de la cual se obtuvieron los derivados etopósido y tenipósido, utilizados para el tratamiento de diferentes tipos de linfomas y leucemias12. También se han reportado estilbenos como las combrestatinas (fig. 2), aisladas de la especie vegetal Combretum caffrum (Combretaceae) donde a partir de la combrestatina A4 se obtuvo el análogo combrestatina A4-fosfato que es soluble en agua y que mostró tener actividad promisoria contra el cáncer de tiroides en los ensayos clínicos preliminares18.
; estilbeno (combrestatina A4, Combrestatina A4P), y flavonoide (quercetina, genisteína, rohitukina, flavopiridol, fenoxodiol, P276-00).")
Compuestos fenólicos bioactivos contra diferentes tipos de cáncer. De tipo lignano (ácido nordihidroguayarético, podofilotoxina, etopósido, tenipósido); estilbeno (combrestatina A4, Combrestatina A4P), y flavonoide (quercetina, genisteína, rohitukina, flavopiridol, fenoxodiol, P276-00).
Los compuestos de tipo flavonoide (fig. 2) que se encuentran presentes en la mayoría de granos, verduras y frutas, están jugando un papel importante en el descubrimiento de nuevos fármacos menos tóxicos, debido a sus efectos protectores frente a algunos tipos de cáncer; hecho que los ha convertido en agentes potenciales para el tratamiento de esta enfermedad11. Aunque algunos flavonoides han comenzado a ser evaluados en estudios clínicos como agentes preventivos, tal vez los más estudiados para el tratamiento de algunos tipos de cáncer de mama, próstata y vejiga son la quercetina, genisteina y rohitukina9,19. Las estructuras químicas de algunos flavonoides han servido de punto de partida para el desarrollo de los agentes terapéuticos flavopiridol, fenoxodiol y el análogo P276-00 (fig. 2), que se encuentran en estudios clínicos contra una amplia gama de tumores, incluidos linfomas y leucemias20.
Tal vez los agentes quimioterapéuticos derivados de productos naturales vegetales más conocidos y utilizados en el tratamiento de diferentes tipos de cáncer son los taxanos21–23 (fig. 3), representados principalmente por el paclitaxel, aislado de la especie vegetal Taxus brevifolia (Taxaceae) y conocido inicialmente como taxol. A partir de esta estructura química se partió para la síntesis del docetaxel y ortataxel (fig. 3), que mostraron ser más activos contra cáncer de seno, ovario y pulmón. Aunque se encuentran 23 taxanos diferentes en estudios preclínicos24,25 y su actividad en el tratamiento del cáncer ha sido de gran ayuda, en la familia Anonáceae se han reportado compuestos de tipo acetogenina como la bullatacina (fig. 3), que mostró ser 300 veces más activa que el taxol cuando se le administró a ratones, a los que se les había inducido un tumor con células L-1210 (cáncer de leucemia). Los ratones tratados con taxol perdieron un 10% de su peso, mientras que los tratados con bullatacina aumentaron su peso en un 5%, lo que sugiere potencialmente que es menos tóxica que el taxol. Actualmente se están estudiando más de 150 tipos de acetogeninas en diferentes líneas celulares de cáncer26,27.
, Anonaceae (bullatacina) y análogos sintéticos (docetaxel, ortataxel).")
También se han reportado algunos compuestos de núcleo quinona (fig. 4) como la β-lapachona aislada del árbol de lapacho Tabebuia avellanedae (Bignoniaceae), la embelina de la especie Embelia ribes (Myrsinaceae) utilizada ampliamente en la medicina tradicional China, y la timoquinona28 de la especie Nigella sativa (Ranunculaceae), los cuales se encuentran en diferentes fases de estudio clínico29 para el tratamiento de algunos tipos de cáncer de colon, ovario, pulmón, próstata y mama.
; amida (piperina, piperlongumina); Oxigenados (alcohol perilílico, dilapiol, curcumina), y análogos sintéticos (NSC295156, NSC698249).")
Compuestos anticancerígenos en diferentes fases de estudio clínico aislados de plantas medicinales. De tipo quinona (β-lapachona, timoquinona, embelina, iriasferina A, SAN5201); amida (piperina, piperlongumina); Oxigenados (alcohol perilílico, dilapiol, curcumina), y análogos sintéticos (NSC295156, NSC698249).
Otros compuestos que son interesantes debido a su simplicidad estructural (fig. 4), gran actividad frente a distintos tipos de cáncer y baja toxicidad, son los análogos sintéticos NSC 29515630, NSC 69824931 y compuestos naturales como la curcumina32, el alcohol perilílico24, el dilapiol33, la piperina34 y la piperlongumina35, siendo estos tres últimos compuestos aislados de diferentes especies del género Piper (Piperaceae). Se ha demostrado que en líneas celulares de cáncer de mama MDA-MB-231, el dilapiol induce la producción de especies reactivas de oxígeno a nivel intracelular, generando la liberación de Citocromo C desde la mitocondria para que este, a su vez, active la vía de muerte celular a través de la producción de caspasas, especialmente la caspasa 3. La piperina, que es obtenida de los frutos de la especie Piper nigrum (Piperaceae) y que es el compuesto responsable del sabor picante de la pimienta, es capaz de potencializar el efecto citotóxico de fármacos como la mitoxantrona y doxorrubicina sobre las líneas celulares MCF7 (cáncer de mama) y A549 (cáncer de pulmón) que se han vuelto resistentes a este tipo de tratamientos36. La piperlongumina aislada de la especie vegetal Piper longum (Piperaceae) y la cual es utilizada ampliamente en la medicina Ayurvédica India, ha demostrado su gran potencial como agente antitumoral, induciendo apoptosis en 13 líneas celulares diferentes que incluyen cáncer de colon, mama, páncreas, óseo, piel y pulmón, sin causar toxicidad en las células sanas37. Adicionalmente, en ensayos realizados in vivo, la piperlongumina logró disminuir de forma considerable el tamaño de los tumores inducidos en ratones con líneas celulares de cáncer MDA-MB-436 (mama), A549 (pulmón) y EJ (vejiga), inclusive en algunos casos de forma más efectiva que en los tumores tratados con paclitaxel o cisplatino.
Mecanismos de acción y quimiorresistenciaLos fármacos antineoplásicos derivados de fuentes vegetales se caracterizan en su gran mayoría por ser agentes antimitóticos, que inducen la muerte de las células cancerosas generalmente cuando se encuentran en el proceso de mitosis38. Esta es la razón principal del porqué afectan también a las células sanas, induciendo daños sobre cualquier célula del cuerpo que se encuentre en proceso de división, lo que genera una serie de efectos secundarios no deseados en la mayoría de los tratamientos con quimioterapia. Por ejemplo, la camptotecina y sus derivados comerciales Topotecan e Irinotecan actúan sobre las topoisomerasas I y II, que son las enzimas encargadas de liberar las tensiones causadas por los enrollamientos del ADN durante su proceso de transcripción, reparación y replicación, haciendo irreversible la regeneración de la cadena de ADN una vez que este ha sido separado para replicarse o liberar tensión39,40.
Los alcaloides de la vinca, las combrestatinas y sus derivados, evitan que sea completada la fase de mitosis en la célula inhibiendo la formación de los microtúbulos, que son las estructuras responsables de separar el material cromosómico y distribuirlo equitativamente entre las dos células hijas. Esta inhibición en la formación del huso mitótico se debe a que estos compuestos se unen a la proteína tubulina e impiden su polimerización durante la formación de los microtúbulos41. Tal vez las combrestatinas hacen parte de los compuestos citotóxicos de los cuales se tiene mayor conocimiento a nivel de su mecanismo de acción, ya que adicionalmente al efecto antimitótico a nivel celular también puede tener un efecto antiangiogénico y antivascular a nivel del tumor42–44, inhibiendo la formación de nuevos vasos sanguíneos y uniéndose a la tubulina celular endotelial de las células que revisten los vasos sanguíneos de los tumores, las cuales generan cambios morfológicos en las paredes vasculares e impiden el flujo de sangre hacia el tumor, para evitar la llegada de nutrientes y oxígeno, lo que induce necrosis a causa de hipoxia e inanición45–47. Estos fármacos son conocidos como agentes bloqueadores vasculares o VDAs48 (por sus siglas en inglés Vascular Disrupting Agents) y están siendo estudiados por el Instituto Nacional del Cáncer en Estados Unidos para el tratamiento de tumores sólidos de rápida evolución, como el fármaco Crolibulin49 utilizado en el cáncer anaplásico de tiroides, o por compañías farmacéuticas como Oxigene para el tratamiento de cáncer de ovario y tumores neuroendocrinos gastrointestinales como es el caso del Fosbrestabulin50,51.
Los lignanos como la podofilotoxina y sus derivados comerciales Etopósido y Tenipósido ejercen su mecanismo de acción no solo afectando a la Topoisomerasa II sino que también se unen a la tubulina, evitando tanto la formación de microtúbulos como la regeneración del ADN después de su replicación. Los taxanos al igual que la podofilotoxina actúan a nivel de los microtúbulos, pero en lugar de inhibir su formación a partir de la polimerización de la tubulina, tienen una gran capacidad para estabilizar sus estructuras e impedir su desacoplamiento, generando un desbalance en la célula que finalmente terminará en apoptosis41.
Otros compuestos como la embelina, la timoquinona y la piperlongumina pueden actuar como antagonistas de las proteínas IAP52, quienes se encargan de controlar a las caspasas, que son las principales proteínas encargadas de ejecutar esta forma de muerte celular programada. Estos compuestos impiden que la proteína XIAP se una a las caspasas, dejando libre las caspasas efectoras de apoptosis como lo son las caspasas 3 y 7, desencadenando en la célula el proceso de muerte celular programada. Por ejemplo, la embelina es efectiva sola o en combinación con otros fármacos sobre células cancerosas que presenten una sobreexpresión de la proteína XIAP, incluso en células PC-3 (cáncer de próstata)53 y en células H460 (cáncer de pulmón) cuando es administrada en combinación con el cisplatino, medicamento que es ampliamente utilizado en quimioterapias al que las células H460 son resistentes54.
Esta quimiorresistencia o resistencia a la quimioterapia ha sido asociada a muchos factores, entre los que se encuentran: (i) la disminución en la captación de algunos fármacos solubles en agua como por ejemplo el cisplatino, análogos de nucleósidos, antagonistas de ácido fólico y (ii) a los diferentes cambios generados en la célula que afectan directamente la capacidad citotóxica de los fármacos, como alteraciones a nivel citoplasmático, cambios en el ciclo celular, aumento en la reparación del ADN y disminución de apoptosis55. Dentro de estos cambios a nivel celular que pueden generar multirresistencia, se puede encontrar un aumento o sobreexpresión de algunas proteínas, como las proteínas IAP36, las cuales además de evitar que la célula entre en apoptosis, contribuyen también a la progresión del tumor56. Las proteínas IAP se encuentran a nivel citoplasmático en la célula, son encargadas de reclutar e inhibir factores proapoptóticos y se pueden considerar como los últimos salvavidas que tiene la célula justo antes de entrar en apoptosis, ya que inhiben la actividad enzimática de las caspasas efectoras llevándolas a su degradación por ubiquitinación 8,57. Este fenómeno se traduce finalmente en que la célula no muere y continúa con sus funciones de división celular de forma descontrolada convirtiéndose así en una célula cancerosa.
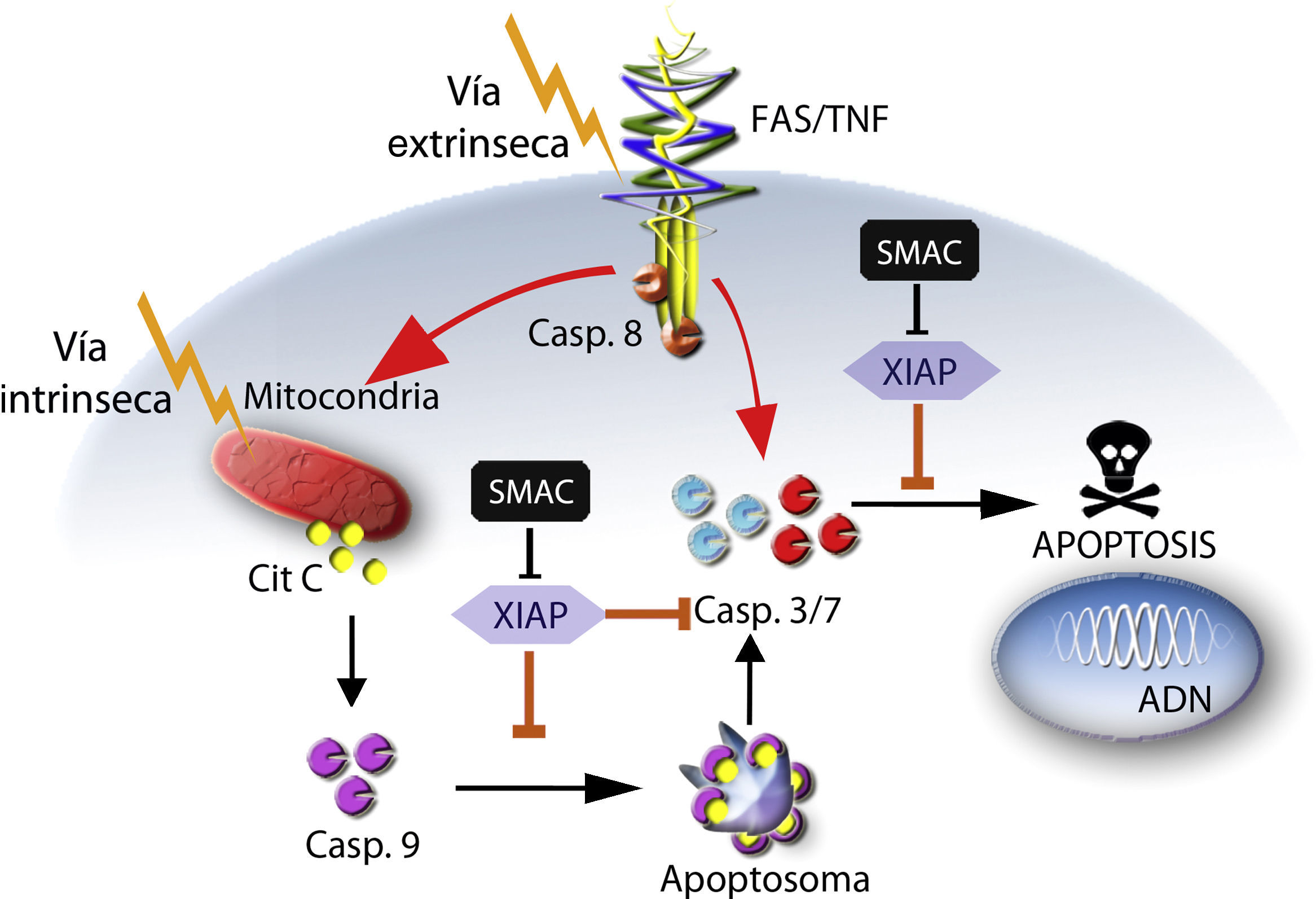
Apoptosis y proteínas inhibidoras de apoptosisLa apoptosis es una forma de muerte celular caracterizada por varios cambios morfológicos, que incluyen condensación de la cromatina, fragmentación nuclear, pliegues en la membrana plasmática, desensamble del citoesqueleto y contracción celular58. Esta forma de muerte celular programada es ejecutada por las caspasas, a través de las cuales la célula realiza de forma autorregulada un proceso interno de destrucción de sus proteínas, citoesqueleto, organelos, membrana nuclear y ADN, agrupando todo el material celular en pequeños cuerpos denominados cuerpos apoptóticos, que posteriormente son eliminados por fagocitosis, sin generar un proceso inflamatorio59. Este proceso de muerte celular programada puede activarse de forma general a través de dos vías: una vía intrínseca y una extrínseca, que dependen directamente del origen del estímulo de muerte60 (fig. 5).
, activación de caspasa 9 (Casp. 9), formación de apoptosoma y posterior inducción de apoptosis a través de la caspasa 3 (Casp. 3); vía extrínseca a través de receptores de membrana asociados a dominios de muerte FasL o TNF-R1, activación de la caspasa 8 (Casp. 8), y posterior inducción de apoptosis a través de la caspasa 3.")
Vías de activación de apoptosis: vía Intrínseca a través de la mitocondria, mediante la liberación de citocromo C (Cit C), activación de caspasa 9 (Casp. 9), formación de apoptosoma y posterior inducción de apoptosis a través de la caspasa 3 (Casp. 3); vía extrínseca a través de receptores de membrana asociados a dominios de muerte FasL o TNF-R1, activación de la caspasa 8 (Casp. 8), y posterior inducción de apoptosis a través de la caspasa 3.
La vía intrínseca es desencadenada en respuesta a una amplia variedad de estímulos que son generados dentro de la célula, tales como activación de oncogenes o daño del ADN. Esta vía es mediada desde la mitocondria, que libera diversas proteínas proapoptóticas hacia el citoplasma celular en respuesta a los estímulos apoptóticos. Algunas de estas proteínas son: el citocromo C, Smac/Diablo (por sus siglas en inglés, Second Mitochondria-derived Activator of Caspase/Direct Inhibitor of Apoptosis-Binding protein with Low pI), AIF (factor inductor de apoptosis), EndoG (endonucleasa G), proteínas de la familia Bcl-2 (por sus siglas en inglés, B-cell lymphoma 2) como Bcl-2, Bcl-XL y Omi/HTRA2 (por sus siglas en inglés, High Temperature Requirement A2)61. La más importante de estas proteínas proapoptóticas es el citocromo C, el cual se une y activa a la proteína APAF-1 (por sus siglas en inglés, Apoptotic Protease Activating Factor-1) en el citoplasma, lo que promueve la formación del apoptosoma, que media la activación autocatalítica de caspasa 9 que a su vez activa a la principal caspasa efectora, la caspasa 356. La vía extrínseca es mediada por la activación de receptores de muerte como Fas o TNF (por sus siglas en inglés, Tumor Necrosis Factor), a través de la unión de un ligando de muerte asociado a su respectivo receptor en la superficie celular. Esta unión conduce a la formación de un complejo ligando-receptor, que seguidamente recluta factores citosólicos, tales como FADD (por sus siglas en inglés, Fas-Associated Death Domain) y caspasa 8, para activar posteriormente a las caspasas efectoras 3 y 756.
Sin embargo, estos procesos de activación de caspasas tanto intrínsecos como extrínsecos que finalizan en apoptosis, se ven interrumpidos o afectados cuando en el citoplasma celular están presentes las proteínas IAP, las cuales se caracterizan por poseer de uno a tres dominios BIR (dominios Repetidos IAP de Baculovirus) capaces de inhibir la activación enzimática de las caspasas. Aunque se conocen ocho proteínas diferentes inhibidoras de apoptosis (NAIP, cIAP1, cIAP2, XIAP, ILP2, livin, survivin y BRUCE)62, las caspasas 3, 7 y 9 son objeto de inhibición principalmente por XIAP y en menor proporción por c-IAP1, c-IAP2 y NAIP63,64. De estas proteínas, XIAP ha sido la más estudiada, se compone de tres dominios BIR (BIR1, BIR2 y BIR3) y un dominio RING (por sus siglas en inglés, Really Interesting New Gene) capaz de unirse a enzimas de ubiquitinación. El papel crucial que juega XIAP en el mecanismo de regulación de la apoptosis, se da principalmente a través de sus dominios BIR2 y BIR3, donde BIR3 inhibe la caspasa 9 y evita la formación del apoptosoma, mientras que la región de unión entre sus dominios BIR1 y BIR2 inhibe las caspasas efectoras 3 y 7 evitando que la célula pueda entrar en un proceso de apoptosis65.
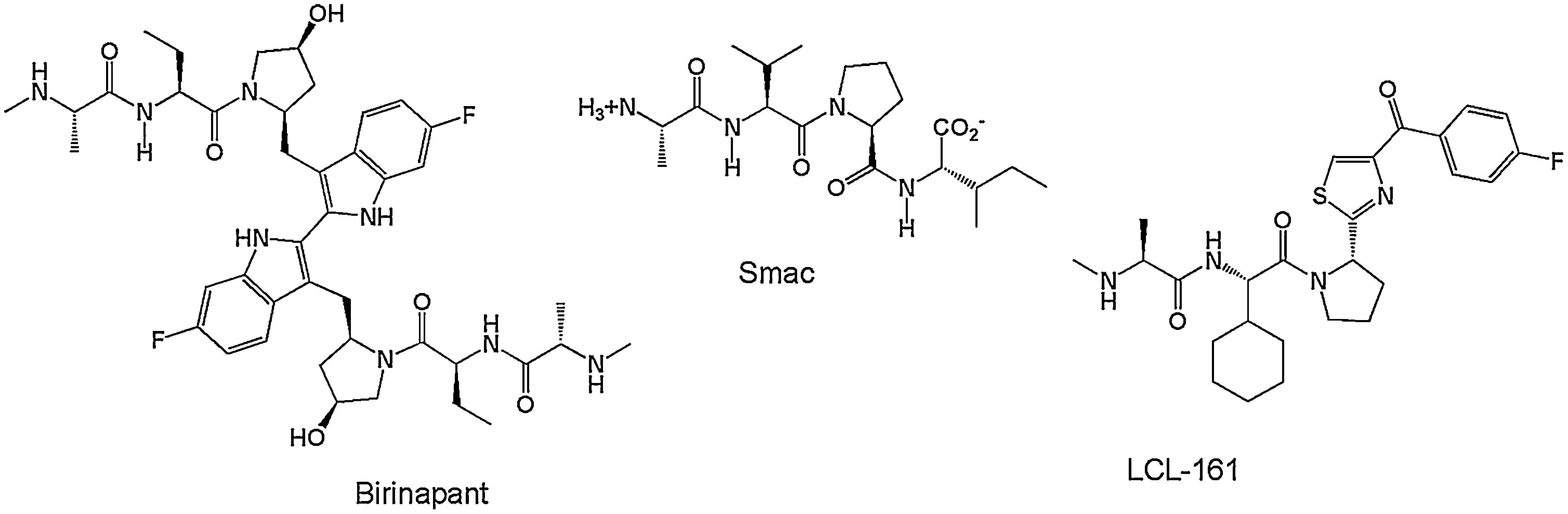
Por otro lado, para que en una célula ocurra el proceso contrario, es decir, que pueda entrar en proceso de muerte celular programada, XIAP debe mantenerse controlada y esta función es realizada por la proteína proapoptótica Smac/Diablo que es liberada por la mitocondria. Smac/Diablo actúa como un antagonista endógeno de XIAP, ya que se une a sus dominios BIR para generar cambios conformacionales que activan la degradación por ubiquitinación de XIAP y evitar así la interacción entre XIAP y las caspasas. Se ha determinado que Smac/Diablo se une fuertemente al dominio BIR3 de XIAP a través del tetrapéptido AVPI (Alanina-Valina-Prolina-Isoleucina)66 (fig. 6) del extremo amino terminal de Smac/Diablo, dejando libres las caspasas activas 3, 7 y 9 para que la célula pueda entrar en proceso de apoptosis67,68. Este tetrapéptido ha servido de punto de partida para realizar la síntesis de otros péptidos similares69–72 con el fin de que actúen como inhibidores de las IAP y promuevan la inducción de muerte celular selectiva frente a líneas celulares tumorales73, como es el caso de los análogos sintéticos Birinapant y LCL-161 que se encuentran en ensayos clínicos contra diferentes tipos de tumores sólidos (fig. 7)74. Sin embargo, estos péptidos imitadores de Smac/Diablo pueden llegar a tener altos costos de síntesis y problemas de solubilidad, permeabilidad y estabilidad dentro la célula.
. Los residuos de unión importantes de BIR3 se resaltan en azul (interacción con AVPI) y rojo (interacción con la cadena lateral de la embelina).")

A diferencia de los péptidos sintéticos, los productos naturales antagonistas de XIAP, como la embelina, tienen mayor facilidad de permear la célula debido al carácter lipofílico que le brinda la cadena lateral de su estructura química. Esta característica apolar, es precisamente la que le permite interactuar con los residuos del bolsillo hidrofóbico de BIR3, tal y como se ha demostrado experimentalmente a través de la interacción con los residuos triptófano-323 y tirosina-324 del dominio BIR3 de XIAP, los cuales también se ha demostrado que interactúan con el residuo AVPI de Smac/Diablo (fig. 6)75,76. Sin embargo, a pesar de que la cadena alquílica lateral de la embelina le brinda carácter lipofílico y facilita su permeabilidad hacia el interior de la célula, también genera algunas limitaciones de solubilidad en medios acuosos debido a su baja polaridad. La embelina se ha administrado por vía oral en dosis de 10mg a 3g/kg en ratas y ratones sin mostrar ningún efecto tóxico77, por lo que algunos grupos de investigación están tratando de solucionar los problemas de solubilidad de la embelina de la misma forma que hace algunos años lo hicieron con las combrestatinas, a través de la síntesis de sales derivadas que sean solubles en agua78.
La piperlongumina y el alcohol perilílico (fig. 4) también han mostrado tener actividad antagonista sobre la proteína XIAP, en especial la piperlongumina que actúa de forma específica sobre distintos tipos de cáncer, sin afectar de forma considerable las células sanas. Su mecanismo no solo involucra la disminución en los niveles de proteínas antiapoptóticas como XIAP y Bcl-2, también se ha demostrado que causa un aumento selectivo en la cantidad de especies reactivas de oxígeno a nivel intracelular, afectando a enzimas de respuesta al estrés oxidativo tales como la GSTP1 y la CBR179. Estas enzimas, posiblemente, también estén involucradas en el mecanismo de acción de la embelina, debido a su estrecha relación estructural con compuestos similares como el SAN5201, identificado como el farmacóforo responsable de la actividad citotóxica de la irisferina A (fig. 4), aislada de las hojas de la especie Iris pseudacorus (Iridaceae). El compuesto SAN5201 y una serie de 11 análogos similares con cadenas alquílicas entre 12 y 17 átomos de carbono, fueron capaces de inducir fuertemente la formación de radicales libres de forma selectiva en las líneas celulares de cáncer A549 (pulmón) y HCT-116 (colon), proceso en el que se ha determinado que la cadena alquílica juega un papel crucial en la generación de radicales libres y citotoxicidad selectiva a células tumorales80.
Aunque no se ha establecido una relación simultánea entre la disminución de los niveles de XIAP, el aumento en las especies reactivas de oxígeno y la disminución en la actividad enzimática de la GSTP1 y la CBR1, es posible que el mecanismo de acción de estos compuestos se relacione directamente con la activación de estos tres procesos proapoptóticos, lo cual aún falta por demostrar experimentalmente. Estos compuestos de estructuras relativamente sencillas a diferencia de la primera generación de medicamentos contra el cáncer donde se utilizaban moléculas estructuralmente complejas y el principal objetivo era generar toxicidad en la célula, están abriendo la puerta a nuevas investigaciones, enfocadas en mecanismos de acción antes desconocidos y que pueden resultar en la obtención de fármacos más selectivos, accesibles y menos tóxicos que los utilizados en la actualidad.
Finalmente, se concluye que la baja especificidad de los tratamientos terapéuticos actuales hacia algunos tipos de cáncer se ve reflejada en la rápida generación de resistencia a los medicamentos y al deterioro de la calidad de vida de los pacientes que padecen esta enfermedad. Las proteínas IAP se han convertido en una de las dianas terapéuticas específicas más importantes en la búsqueda de nuevos fármacos contra el cáncer, bien sea a través del desarrollo de péptidos imitadores de Smac/Diablo o la búsqueda de compuestos naturales o sintéticos que tengan efecto antagonista sobre la proteína XIAP. Los compuestos como la embelina y la piperlongumina, aislados de especies vegetales que desde hace mucho tiempo se vienen utilizando en la medicina tradicional de India y China; basan su mecanismo de acción en causar alteraciones sobre las proteínas que están involucradas directamente en el mecanismo de inducción de apoptosis en células tumorales y abren las puertas a la búsqueda de tratamientos médicos más selectivos, sin que se vean afectadas también las células sanas. Aunque no se ha demostrado una relación directa entre el aumento de especies reactivas de oxígeno y un efecto antagonista sobre las proteínas inhibidoras de apoptosis, es apenas evidente que la cadena alquílica lateral de compuestos como la embelina y el SAN5201 cumple un papel fundamental en la inducción de apoptosis, ya que por un lado disminuye los niveles de la proteína XIAP y, por otro, aumenta la producción intracelular de radicales libres de forma selectiva en células tumorales. La naturaleza ha sido la fuente principal de los medicamentos descubiertos durante el siglo pasado y muchos de los fármacos útiles se han desarrollado a partir de fuentes naturales, en especial de las plantas; por lo tanto, es de esperar que la naturaleza siga siendo una fuente importante en el descubrimiento de nuevos medicamentos en el futuro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
