



Los leiomiosarcomas pueden originarse en la mayor parte de los órganos desarrollados fuera del sistema nervioso central. Se han documentado casos de leiomiosarcomas de origen: intestinal, mesentérico, uterino, retroperitoneal, de tejidos blandos y vascular. Sin embargo, los casos de leiomiosarcomas primarios de ovario son extremadamente infrecuentes y pocos casos han sido descritos en la literatura. Se cree que estos leiomiosarcomas se originan en el músculo liso alrededor de los folículos o a partir de remanentes del conducto de Wolff a partir de las paredes de la vasculatura existente en el parénquima ovárico. La mayor parte de los casos se presentan en pacientes postmenopáusicas y con un pronóstico a corto plazo desfavorable. El pilar del tratamiento continúa siendo la citorreducción tumoral completa obteniendo márgenes quirúrgicos negativos con miras a disminuir el potencial de recidiva. El beneficio de la utilización de quimioterapia adyuvante como parte del tratamiento de esta patología sigue siendo incierto.
Leiomyosarcomas may originate in most of the organs developed outside the central nervous system. There are documented cases of leiomyosarcomas of intestinal, mesenteric, uterine, retroperitoneal, and of soft and vascular tissue origin. However, cases of primary ovarian leiomyosarcoma are extremely rare, with few cases reported in the international literature. Leiomyosarcomas are believed to be those that originate from the walls of existing vasculature in ovarian parenchyma, in the smooth muscle around the follicles, or from remnants of the Wolff duct. Most cases occur in post-menopausal patients, and have an unfavourable prognosis in the short term. The mainstay of treatment remains the complete tumour debulking, with negative surgical margins in order to reduce the potential for recurrence. The benefit of the use of adjuvant chemotherapy as part of treatment of this condition remains uncertain.
El leiomiosarcoma primario del ovario es un tumor extremadamente raro, representa menos del 1% de todos los tumores malignos del ovario1–3. En la actualidad no existen datos epidemiológicos exactos en relación con su tasa de presentación dado que la mayor parte de la información disponible proviene de reportes que hasta la fecha no superan los 70 casos en la literatura internacional y ninguno publicado en la literatura nacional2. La mayoría de estos tumores se presentan en mujeres postmenopáusicas con algunos casos excepcionales registrados en mujeres jóvenes. Generalmente es un tumor de tamaño variable, unilateral y de rápido crecimiento4. En la mayoría de los casos presenta un comportamiento agresivo con un pronóstico desfavorable y con una mortalidad significativa dentro de los dos años siguientes al tratamiento oncológico inicial5.
A continuación se presenta el caso clínico de una paciente postmenopáusica con diagnóstico de leiomiosarcoma de origen ovárico estadio FIGO IC 3 según la International Federation of Gynecologists and Obstetricians, llevado a manejo quirúrgico primario y a tratamiento adyuvante con quimioterapia, cuya revisión tiene una significativa relevancia ante la ausencia de otros casos reportados en la literatura nacional y con el que se pretende aportar datos útiles acerca de la caracterización epidemiológica y del comportamiento de esta patología inusual.
Presentación de casoPaciente femenina de 61 años, con antecedente de 11 gestaciones, 10 partos vaginales y 1 aborto, sin antecedentes patológicos de relevancia, quien consulta por cuadro de tres meses de evolución de sangrado postmenopáusico, asociado a percepción de pérdida de peso. Al examen físico con hallazgo de masa abdominal de aproximadamente 20cm, móvil, de localización en hipogastrio y mesogastrio.
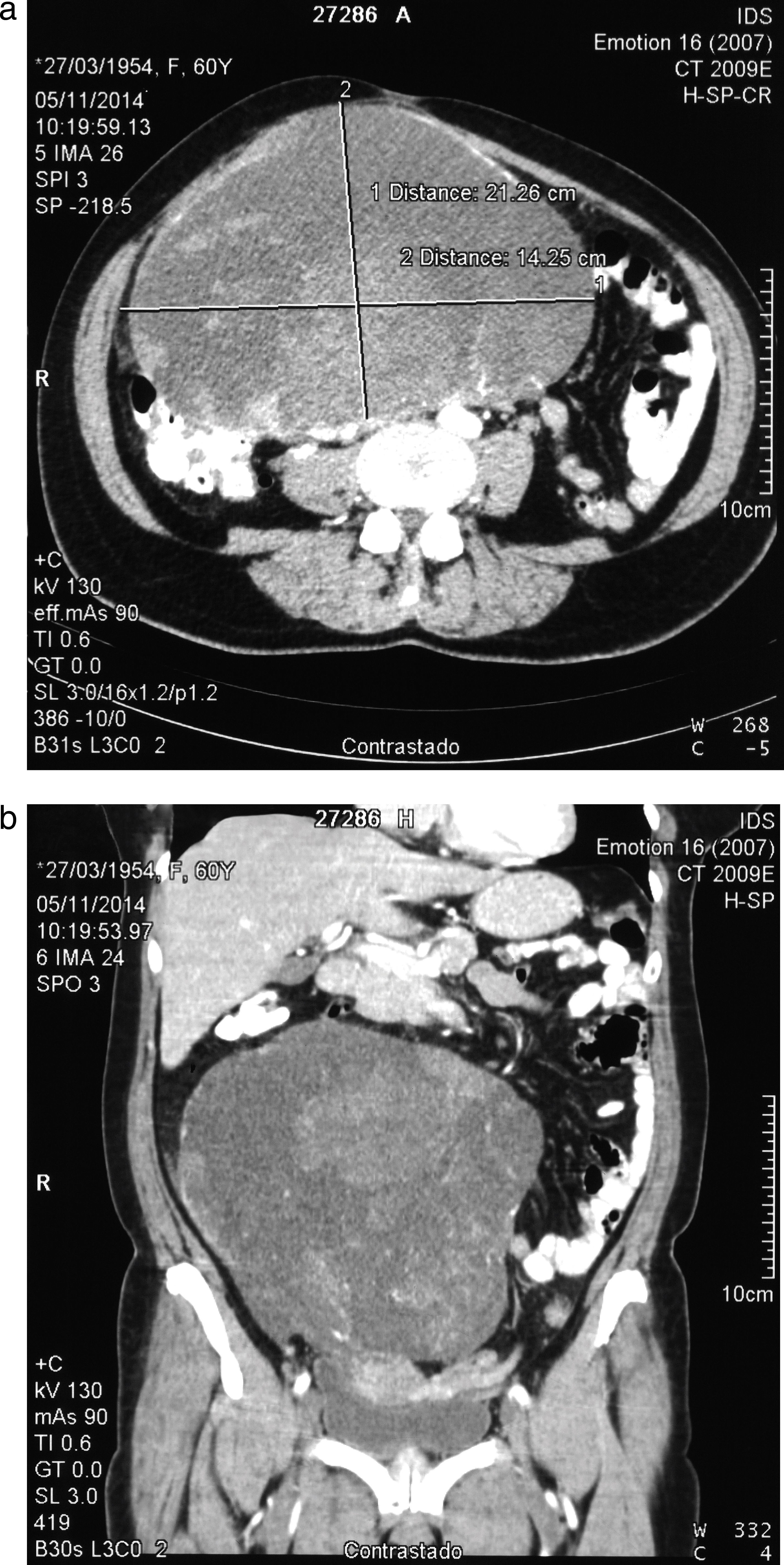
Se realizan estudios de laboratorio y de imagenología complementarios; en la tomografía axial computarizada de abdomen y pelvis se identificó lesión ocupante de espacio dependiente de ovario derecho de 20×14cm de apariencia sólido quística (fig. 1). Los hallazgos en la tomografía de tórax fueron normales, la biopsia endometrial realizada en consultorio fue negativa para compromiso tumoral endometrial y el Ca 125: 350.
 corte axial que evidencia masa de 20×14cm, con áreas de degeneración sólido quística, (b) reconstrucción coronal que evidencia la masa descrita dependiente de anexo derecho y su relación con las demás estructuras abdominales.")
Tomografía axial computarizada de abdomen con contraste (a) corte axial que evidencia masa de 20×14cm, con áreas de degeneración sólido quística, (b) reconstrucción coronal que evidencia la masa descrita dependiente de anexo derecho y su relación con las demás estructuras abdominales.
Teniendo en cuenta la presentación clínica descrita, los hallazgos en estudios de imagenología y marcador tumoral, se realiza diagnóstico presuntivo de tumor de origen ovárico por lo que se plantea llevar a cirugía de citorreducción primaria.
Dentro de los hallazgos intraoperatorios se evidenció líquido ascítico cetrino (aproximadamente 300 cc) y lesión tumoral de 32×28cm que comprometía el anexo derecho con excresencias en su superficie, sin adherencias a estructuras vecinas. El resto de órganos (útero, recto sigmoide, asas intestinales delgadas y gruesas, hígado, bazo, superficies peritoneales de cúpulas diafragmáticas, apéndice cecal, estómago, cadenas ganglionares pélvicas y para aórticas) presentaban aspecto normal.
Se realizó citorreducción completa de la enfermedad, incluyendo dentro de los procedimientos realizados: resección de tumor descrito, histerectomía abdominal total, salpingooforectomía izquierda, omentectomía, linfadenectomía pélvica bilateral y paraaórtica. La paciente presentó evolución satisfactoria en su postoperatorio inmediato, obteniendo egreso hospitalario a los dos días de la intervención quirúrgica.
El estudio histológico de las muestras enviadas demuestran líquido ascítico y lavados peritoneales positivos para malignidad. Macroscópicamente el tumor fue descrito como masa de 30×28×18cm con peso de 3.800 gr. Al corte del espécimen se apreció multiloculado, con contenido líquido y mucinoso, con áreas de necrosis en su interior y con presencia de nódulos tumorales sobre la superficie de la cápsula.
Los hallazgos microscópicos corresponden a una lesión neoplásica maligna constituida por: células de aspecto estromal, en su mayoría fusiformes, con un patrón arremolinado y en disposición fascicular; con atipia celular marcada; áreas de necrosis hemorrágica y mitosis atípica mayor a 20 en 10 campos de alto poder, compatible con tumor fuso celular maligno de alto grado.
Los hallazgos del estudio inmunohistoquímico son: positivos para caldesmón, actina de músculo liso, desmina, citoqueratina (AE1/AE3), receptores de progesterona (focal), WT-1 débil, S100, con índice de proliferación (Ki-67) del 100%, sin evidencia de invasión linfovascular; y hallazgos negativos para cromogranina, sinaptofisina, EMA, CD-99, inhibina, calretinina; inmunofenotipo que favorece leiomiosarcoma (fig. 2).
, (b), (c) Lesión neoplásica maligna constituida por células de aspecto estromal, fusiformes con disposición fascicular, atipia celular marcada, áreas de necrosis hemorrágica y mitosis atípica mayor a 20 en 10 campos de alto poder. Inmunohistoquímica positiva para (d) desmina; (e) actina de músculo liso; (f) S100 (nuclear); (g) índice de proliferación (Ki-67) del 100%; (h) citoqueratina coctel (AE1/AE3) y; (i) receptores de progesterona.")
(a), (b), (c) Lesión neoplásica maligna constituida por células de aspecto estromal, fusiformes con disposición fascicular, atipia celular marcada, áreas de necrosis hemorrágica y mitosis atípica mayor a 20 en 10 campos de alto poder. Inmunohistoquímica positiva para (d) desmina; (e) actina de músculo liso; (f) S100 (nuclear); (g) índice de proliferación (Ki-67) del 100%; (h) citoqueratina coctel (AE1/AE3) y; (i) receptores de progesterona.
El estudio patológico del útero y del anexo izquierdo fue negativo para compromiso por tumor y los ganglios linfáticos pélvicos y para aórticos se informaron como negativos para malignidad. Con este resultado se clasificó como un leiomiosarcoma dependiente de ovario derecho estadificación IC 3, dada la presencia de líquido ascítico positivo para malignidad y el compromiso tumoral evidenciado en la superficie ovárica, según FIGO. Se consideró candidata a inicio de tratamiento adyuvante con quimioterapia por lo que se administraron 6 ciclos de ifosfamida y doxorrubicina liposomal con adecuada tolerancia.
Veinte meses después de finalizado el tratamiento adyuvante, la paciente se encuentra libre de enfermedad de acuerdo con la valoración clínica, por imágenes diagnósticas y marcadores serológicos.
Se obtuvo consentimiento de la paciente para obtener y presentar las imágenes con objetivos académicos.
DiscusiónLos sarcomas de origen ovárico son extremadamente raros, constituyen un subgrupo de tumores malignos que corresponden a menos del 3% de los tumores de ovario6. La relación de incidencia entre sarcoma de ovario y carcinoma epitelial de ovario se ha establecido en 1 a 40 respectivamente7. La mayor parte de los sarcomas de ovario corresponden a fibrosarcomas, rabdomiosarcomas, sarcomas estromales y menos frecuentemente (menos del 1% de todos los tumores malignos de ovario) a leiomiosarcomas, que a su vez, pueden clasificarse en mixoides o epitelioides1–4.
En cuanto a la edad de presentación, la mayor parte de los casos se diagnostican en pacientes postmenopáusicas. Sin embargo, el rango de edad es muy amplio en los casos registrados pues abarca desde 12 hasta 84 años. La edad promedio de presentación se ha calculado en 53 años1,7. Solo el 15,2% de pacientes que presentan la enfermedad son menores de 40 años y menos del 10% son menores de 30 años8,9.
El origen histológico y la patogénesis de este tumor maligno mesenquimal son todavía inciertos. Se han propuesto múltiples teorías al respecto, dentro de las que se incluyen la degeneración maligna de un leiomioma de ovario o su origen a partir del músculo liso presente en la pared de los vasos sanguíneos, del hilio ovárico, del estroma cortical o del cuerpo lúteo10,11. También se ha considerado su origen a partir de: las inserciones musculares del ligamento ovárico; los remanentes del conducto de Wolff; la presencia de mesénquima ovárico totipotencial, o como resultado de la conversión maligna de un teratoma o la migración de un leiomioma uterino1,2,5–7.
En los casos de las pacientes de 12 años reportados con esta patología, se encontró antecedente de tratamiento con radioterapia por diagnóstico de meduloblastoma lo que podría sugerir como posible factor de riesgo el haber recibido tratamiento radioterápico previo8.
Estos tumores se presentan con síntomas inespecíficos relacionados con: presencia de masa abdominal; dolor abdomino pélvico; trastornos del hábito gastrointestinal y urinario; sensación de plenitud gástrica, o también pueden manifestarse con hemorragia uterina anormal como el caso de la paciente en consideración3,9.Típicamente se presentan como una gran masa unilateral mayor de 10cm 5, siendo un tumor sólido, lobulado, de coloración blanco - pálido a gris - amarillo; puede estar asociada con degeneración quística o con áreas de hemorragia focal y necrosis evidente9,11.
En una revisión de la literatura de 26 casos de leiomiosarcomas de origen ovárico, el estadio de presentación fue variable, encontrándose: el 58% de los casos (15 pacientes) en estadio I, el 15% (4 pacientes) en estadio II y el 27% (7 pacientes) en estadio III9.
El tumor microscópicamente está compuesto por células fusiformes dispuestas en haces entrelazados y fascículos, y presenta una alta celularidad y pleomorfismo nuclear. Existen diferentes variantes como: los leiomiosarcomas mixoides, los leiomosarcomas epitelioides y los leiomiosarcomas con células gigantes osteoclásticas6,11.
Los criterios histológicos usados para evaluar los sarcomas de cuerpo uterino se han homologado al diagnóstico de los leiomiosarcomas de origen ovárico. Dentro de estos se tiene en cuenta: la hipercelularidad; la atipia nuclear; el pleomorfismo; la tasa de miotosis atípica por campo de alto poder, y la necrosis de coagulación. Se considera que el diagnóstico puede hacerse con la presencia en el espécimen estudiado de al menos dos criterios: necrosis por coagulación, atipia celular e índice mitótico mayor a 10 en 10 capos de alto poder8,9. La valoración histológica del tipo leiomiosarcoma mixoide puede ser limitada debido a la baja celularidad y el abundante estroma mixoide, sugiriéndose que el conteo mitótico convencional debe realizarse de forma separada en las áreas sólidas y en las áreas mixoides7.
El diagnóstico diferencial se establece fundamentalmente con: leiomiomas celulares de ovario, neoplasias malignas complejas ováricas con componente de músculo liso y sarcomas metastásicos de origen extraovárico4.
Con respecto a los leiomiomas celulares benignos de ovario de tipo leiomioma mitóticamente activo y especialmente, de tipo leiomioma con núcleos bizarros del ovario, se deben tener en cuenta los factores histológicos nombrados anteriormente para establecer diagnóstico diferencial. El leiomioma mitóticamente activo suele tener una atipia celular menos frecuente, encontrándose menos de 3 mitosis en 10 campos de alto poder. En el caso del leiomioma con núcleos bizarros puede aceptarse hasta 7 mitosis en 10 campos de alto poder5,8,9.
Los marcadores de inmunohistoquímica para los leiomiosarcomas primarios de ovario registrados en la mayor parte de los casos reportados en la literatura9, muestran reactividad positiva a la actina de músculo liso específica (SMA), desmina, vimentina y p53. Y negatividad variable para S-100. Los receptores de estrógeno y progesterona son positivos en el 33 y 50% respectivamente de los leiomiosarcomas primarios de ovario5,8,12.
Para el caso presentado en esta revisión, además de obtenerse positividad para actina de músculo liso y desmina, se utilizaron otros marcadores para tumores de músculo liso como h-caldesmón y contrario a lo registrado en el resto de las revisiones, se obtuvo positividad nuclear para S-100. Asociado a esto se obtuvo reactividad positiva del 100% para Ki-67 y reactividad focal para progesterona.
También debe hacerse diferenciación entre leiomiosarcomas de origen ovárico y los tumores de músculo liso de origen gastrointestinal (GIST) cuyo diagnóstico en la histología habitual sigue siendo un reto, por lo que un perfil de inmunohistoquímica C-kit y CD-34 negativo y desmina positivo puede ayudar a determinar el origen ovárico y no gastrointestinal.
Otros diagnósticos diferenciales incluyen: los teratomas inmaduros con componente leiomiomatoso prominente, el schwannoma maligno, los tumores de cordones sexuales–estromales del ovario, y fibromas y fibrosarcomas8. En este último caso, los fibrosarcomas se diferencian por no tener una importante distribución de células en fascículos, así como por la presencia de un fondo predominante de colágeno. La reactividad a la desmina puede ser negativa o focalmente positiva a diferencia de la positividad difusa de esta que se evidencia en los leiomiosarcomas de ovario9.
Los leiomiosarcomas del ovario son mucho más agresivos que sus tumores homólogos en el cuerpo uterino. En las revisiones de casos de leiomiosarcomas primarios de ovario de pacientes llevadas a seguimiento clínico, se ha encontrado que después del procedimiento quirúrgico inicial, el 71% de los casos, presentan recurrencia de la enfermedad en un intervalo de 19 meses y más de la mitad mueren por la enfermedad en un intervalo de 24 meses. Los sitios de recurrencia más frecuentes son la extensión locorregional en la pelvis y el abdomen en el 38% de los casos, el pulmón en el 14%, el hueso en el 9% y en el hígado, el mediastino y el cerebro cada uno con una tasa de 5% de los casos respectivamente9.
Se considera que el pronóstico de la enfermedad se correlaciona directamente con el estadio de presentación1, de esta forma, la mayor parte de los pacientes que se diagnostican en estadio I permanecen vivos y libres de enfermedad durante un tiempo de seguimiento de 41,7 meses, mientras que todos los pacientes diagnosticados en estadios II y III murieron en un promedio de 14,7 a 24 meses9. Se han propuesto algunos otros factores pronóstico de tipo histológico, dentro de los que se incluyen la actividad mitótica, la actividad proliferativa y la expresión de p53, Ki-67 y bcl-2 5. Pese a esto, no hay datos conclusivos a cerca de su clara influencia en el pronóstico1,4,8.
En el presente caso se confirma que tanto la citorreducción completa de la enfermedad como el estadio en el momento del diagnóstico son factores pronóstico fundamentales, de tal forma que esta paciente con estadio IC3, permanece libre de enfermedad después de 20 meses posteriores a la intervención quirúrgica y para este caso particular, después de haber recibido tratamiento adyuvante con quimioterapia.
No hay estudios prospectivos que hayan concluido recomendaciones sólidas a cerca del manejo de los leiomiosarcomas de origen ovárico y la ausencia de estos datos específicos ha llevado a adaptar algunos principios del tratamiento existentes para leiomiosarcomas de origen uterino13. La primera línea de tratamiento es quirúrgica, realizándose histerectomía total abdominal, salpingooforectomía bilateral y extirpación de las masas tumorales2,3,7. En pacientes en quienes no se ha realizado linfadenectomía ni omentectomía, una segunda intervención en busca de metástasis ocultas es innecesaria debido a la baja probabilidad de que estas se presenten13.
El beneficio de la terapia adyuvante tanto en estadios tempranos como estadios avanzados de la enfermedad no se ha conocido claramente y ninguna modalidad de tratamiento adyuvante, ya sea quimioterapia o radioterapia, ha mostrado ser superior8.
La quimioterapia tiene un papel fundamental en el tratamiento de la enfermedad metastásica irresecable. Si bien, su intención no es curativa, puede retrasar la progresión de la enfermedad sistémica13.
La radioterapia permite mejorar las tasas de control local y puede utilizarse como herramienta con intención paliativa en aquellos casos de enfermedad metastásica1,2.
Por último concluimos, según el caso que presentamos en esta revisión y demás casos e información consultada, que el leiomiosarcoma de origen primario en ovario es un tumor maligno infrecuente y se presenta en mujeres principalmente postmenopáusicas. El comportamiento es agresivo en la mayoría de los casos, presentándose recurrencia local temprana y la muerte antes de los primeros 24 meses. El principal factor pronóstico definido es el estadio de la enfermedad en el momento del diagnóstico. Los pocos casos registrados en la literatura y la ausencia de estudios prospectivos no permiten definir recomendaciones con respecto a la extensión del manejo quirúrgico y la adyuvancia con quimioterapia y radioterapia.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
