


La eriptosis se ha descrito como el proceso de muerte celular programada en el eritrocito antes de la senectud. Puede desencadenarse en situaciones como: el estrés osmótico, el estrés oxidativo, la exposición a metales pesados, entre otros factores. Diversos estudios sugieren que los eritrocitos pueden desempeñar un papel activo en la hemostasia normal o anormal en ciertas condiciones en las que se produce perturbación de la membrana de estas células.
ObjetivoDescribir los mecanismos involucrados en la eriptosis y su estrecha relación con los procesos de adhesión a la pared vascular que conllevan a la enfermedad trombótica.
MétodoSe hizo una revisión narrativa a partir de la literatura encontrada en las bases de datos PubMed y Science Direct utilizando las palabras claves. Se seleccionaron 51 artículos originales, 20 revisiones de la literatura y un estudio de casos, que se ajustaban a las exigencias del objetivo. Se revisaron los resúmenes de forma separada e independiente. Seguidamente se buscaron las publicaciones en el texto completo para la revisión.
ConclusionesLa eriptosis se caracteriza por la disminución del volumen celular, la vesiculación y la translocación del fosfatidil serina hacia la superficie externa de la membrana plasmática. Las alteraciones en la distribución de los fosfolípidos favorecen los procesos de adhesión celular a la pared vascular, conllevando al deterioro de la microcirculación, lo cual puede ocasionar importantes trastornos a nivel cardiovascular. La comprensión y el esclarecimiento de la eriptosis pueden ser esenciales para la búsqueda de nuevas dianas terapéuticas, encaminadas a ofrecer otras alternativas farmacológicas en el tratamiento de la enfermedad cardiovascular.
Eryptosis has been described as programmed process of cellular death in erythrocytes before old age. It can be triggered, among other factors, by situations such as osmotic stress, oxidative stress or exposure to heavy metals. Several studies suggest that erythrocytes can play an active role in normal or abnormal haemostasis in certain conditions where the membrane of these cells is perturbed.
ObjetiveTo describe the mechanisms involved in eryptosis and their close relationship with the processes of adhesion to the vascular wall that entail the thrombotic disease.
MethodsA narrative review was carried out from the literature found in data base PubMed and Science Direct by using the key words. 51 original articles, 20 literature reviews and one case study complying with the requirements were selected. Abstracts were reviewed separately and independently. Complete publications were then located for review.
ConclusionsEryptosis is characterised by the decrease in cell volume, the vesiculation and the translocation of phosphatidylserine towards the outer surface of the plasma membrane. Disorders in the distribution of the phospholipids favour processes of cell adhesion to the vascular wall, causing impairment of microcirculation, which can result in important cardiovascular diseases. Understanding and clarifying eryptosis could be essential for finding new therapeutic targets, aimed at offering other pharmacological alternatives for the treatment of cardiovascular diseases.
Los eritrocitos constituyen la mayoría de los componentes celulares de la sangre. Su participación en la hemostasia y en los procesos trombóticos no está claramente comprendida. No obstante, muchos estudios clínicos han reportado una estrecha relación entre el número de eritrocitos con los tiempos de coagulación y la aparición de enfermedades trombóticas1,2. Por ejemplo, la prolongación de los tiempos de la coagulación puede ser tratada con la transfusión de eritrocitos3. Los pacientes con policitemia vera, a menudo experimentan aumento de trombosis, encontrándose el aumento en el recuento de eritrocitos como un marcador predictivo para su aparición4.
Los eritrocitos están desprovistos del núcleo y las mitocondrias, por tanto, carecen de la maquinaria biológica necesaria para realizar el proceso clásico de muerte celular programada o apoptosis5,6. Sin embargo, investigaciones realizadas sugieren que estas células pueden sufrir un mecanismo similar a la apoptosis antes de su senescencia7, denominado actualmente “eriptosis”8. Por otra parte, diversos reportes indican la relación de este evento con los procesos de adhesión a la pared vascular2,9–11.
Estos hallazgos sugieren que los eritrocitos pueden desempeñar un papel activo en la hemostasia normal o anormal en ciertas condiciones en las que se produce perturbación de la membrana celular de estas células. En esta revisión se describen los mecanismos moleculares involucrados en la eriptosis al igual que la influencia de los fosfolípidos en los procesos de adhesión a la pared vascular, sus implicaciones en la trombosis y otras patologías.
MetodologíaEstrategia de búsqueda de la informaciónFue realizada una búsqueda en las bases de datos, donde se incluyeron diferentes tipos de publicaciones tales como: artículos originales, artículos de revisión y estudio de casos sobre la eriptosis, además, de enfermedades cardiovasculares relacionadas con los eritrocitos. La búsqueda bibliográfica se realizó por medio electrónico en las bases de datos PubMed y Science Direct.
Métodos de revisiónSe evaluaron los títulos de los artículos encontrados. En el caso de relacionarse con el objetivo de la revisión, fueron analizados todos los resúmenes de los artículos identificados con la finalidad de realizar una nueva selección. Finalmente, los artículos seleccionados fueron revisados en su totalidad.
Recopilación y análisis de datosSe identificaron 225 artículos utilizando las palabras claves, de los cuales 72 fueron considerados por su pertinencia a la revisión temática. De éstos, 51 artículos eran artículos originales, 20 revisiones de la literatura y un estudio de casos, que se ajustaban a las exigencias del objetivo. Se revisaron los resúmenes de forma separada e independiente. Seguidamente, se buscaron las publicaciones con el texto completo y se citaron a lo largo de la revisión.
Mecanismos moleculares de la eriptosisLa eriptosis es un mecanismo de muerte programada de eritrocitos en el que se producen alteraciones en la membrana, como externalización de la fosfatidilserina (PS) y generación de microvesículas, que pueden permitir a los eritrocitos participar en los procesos de: coagulación sanguínea, adhesión celular y eliminación de eritrocitos inmaduros2,8. Diversos estudios han descrito la exposición de la fosfatidilserina en la cara externa de la membrana como un marcador de muerte eritrocitaria8,12–15. La exposición de ésta proporciona un sitio de ensamblaje para el complejo enzimático protrombinasa y tenasa, lo cual conduce a la generación de trombina, incremento en la coagulación sanguínea y aumenta la probabilidad de eventos trombóticos16–18. También, se ha descrito que estos eritrocitos se unen con más facilidad a las células endoteliales contribuyendo a una mayor vaso-oclusión19.
La eriptosis es un proceso coordinado de eliminación de células defectuosas, en la cual no hay ruptura de la membrana celular ni liberación del material intracelular. Se caracteriza por la pérdida de la asimetría de la membrana celular y la reorganización de los fosfolípidos14,15. Normalmente, en la membrana plasmática del eritrocito, la esfingomielina y la fosfatidilcolina se encuentran en la cara externa de la bicapa de la membrana, mientras que la fosfatidiletanolamina y la fosfatidilserina se encuentran en el interior16. La exposición de este último a la superficie celular y la generación de microvesículas procoagulantes en los eritrocitos, está mediada principalmente por el aumento de iones de calcio intracelular7,20, con la consecuente activación de la proteína quinasa C (PKC)21.
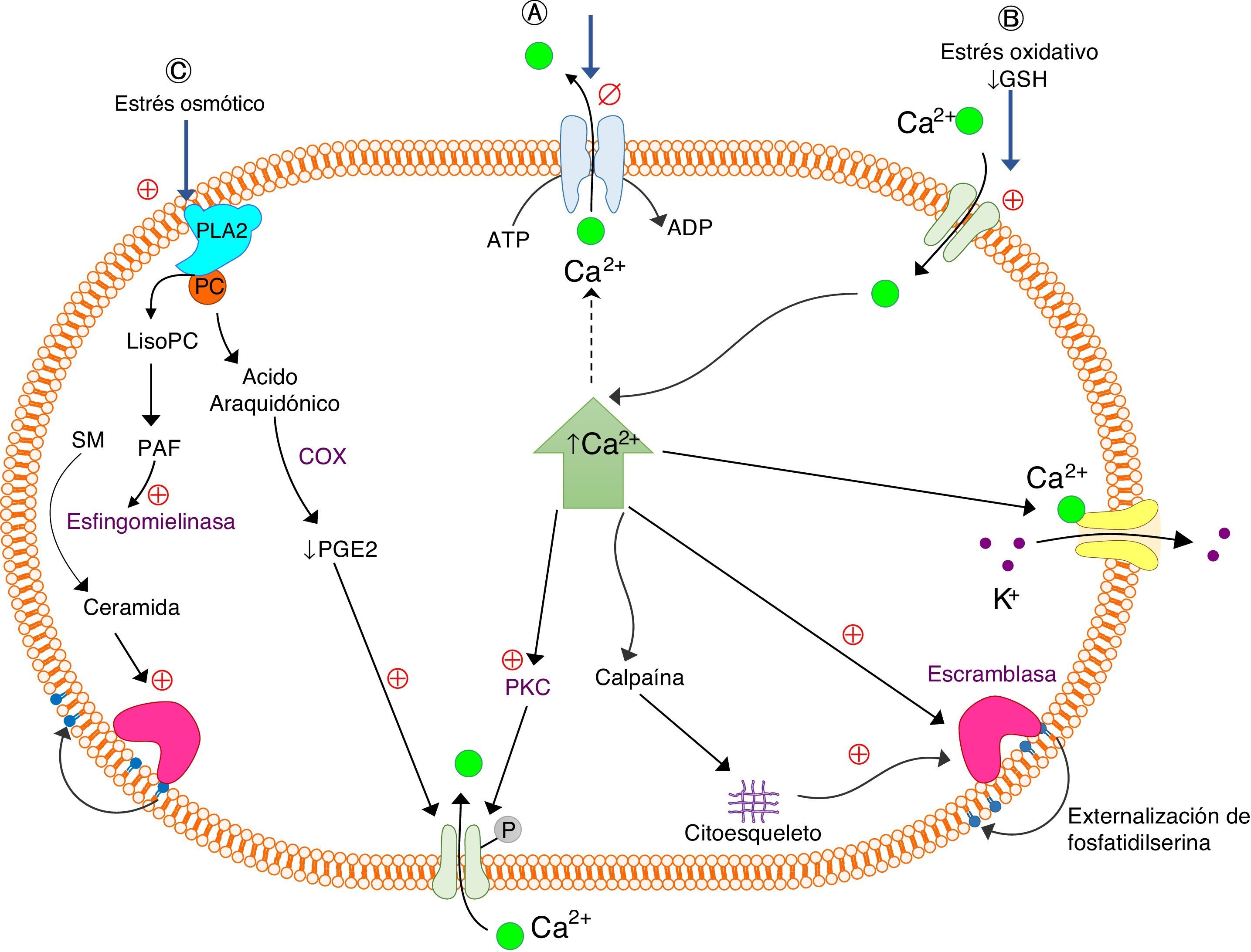
La eriptosis puede ser estimulada principalmente por el estrés osmótico, el oxidativo y el energético (fig. 1)22–24; aunque los patógenos intracelulares, los metales pesados como: el mercurio (Hg), el cadmio (Cd), entre otros, también pueden incidir en dicho proceso13,25–28. El aumento de los niveles de iones del Ca2+ libre intracelular conllevan a la activación de μ-calpaína, una endopeptidasa cisteínica que degrada las proteínas del citoesqueleto; por tanto, se inicia la formación de vesículas en la membrana celular con la consecuente activación de la eriptosis5,6,29. Los eritrocitos cambian su morfogía al perder la simetría de la membrana celular. En un eritrocito que expone la fosfatidilserina en su membrana celular, la forma normal discoide de esta célula puede pasar a diferentes formas que conllevan a la microvesiculación de la membrana plasmática (fig. 2)2,30.
 esquematizados en la figura es el aumento de Ca2+ intracelular, pero cada uno se explica por vías diferentes. A. Estrés energético: disminución de ATP que reduce la acción de la ATPasa de calcio y en consecuencia disminuye la salida de Ca2+. Activación de PKC que fosforila proteínas de la membrana que permiten la entrada de Ca2+. B. Estrés oxidativo: disminución de GSH y activación de los canales catiónicos. C. Estrés osmótico: activación de la PLA2, que libera ácido araquidónico de fosfatidilcolina, para generar PGE2, que activa los canales de Ca2+. Activación de los canales de K+dependientes de Ca2+, con la subsiguiente pérdida de Cl- y H2O, generando contracción en el eritrocito. Los tres procesos esquematizados estimulan la translocación de fosfolípidos de la membrana, produciendo la exposición o externalización de PS por medio de una escramblasa sensible a Ca2+. Glutatión reducido (GSH); fosfolipasa A2 (PLA2); fosfatidilcolina (PC); prostaglandina E2 (PGE2); esfingomielina (SM); factor de agregación plaquetario (PAF); ciclooxigenasa (COX); proteína quinasa dependiente de calcio (PKC); estimulación (⊕); inhibición (∅). Adaptado de Herlax y col. 2011.")
Señalización celular de los procesos de la eriptosis. El estímulo clave para la eriptosis en los tres procesos (A, B y C) esquematizados en la figura es el aumento de Ca2+ intracelular, pero cada uno se explica por vías diferentes. A. Estrés energético: disminución de ATP que reduce la acción de la ATPasa de calcio y en consecuencia disminuye la salida de Ca2+. Activación de PKC que fosforila proteínas de la membrana que permiten la entrada de Ca2+. B. Estrés oxidativo: disminución de GSH y activación de los canales catiónicos. C. Estrés osmótico: activación de la PLA2, que libera ácido araquidónico de fosfatidilcolina, para generar PGE2, que activa los canales de Ca2+. Activación de los canales de K+dependientes de Ca2+, con la subsiguiente pérdida de Cl- y H2O, generando contracción en el eritrocito. Los tres procesos esquematizados estimulan la translocación de fosfolípidos de la membrana, produciendo la exposición o externalización de PS por medio de una escramblasa sensible a Ca2+. Glutatión reducido (GSH); fosfolipasa A2 (PLA2); fosfatidilcolina (PC); prostaglandina E2 (PGE2); esfingomielina (SM); factor de agregación plaquetario (PAF); ciclooxigenasa (COX); proteína quinasa dependiente de calcio (PKC); estimulación (⊕); inhibición (∅). Adaptado de Herlax y col. 2011.
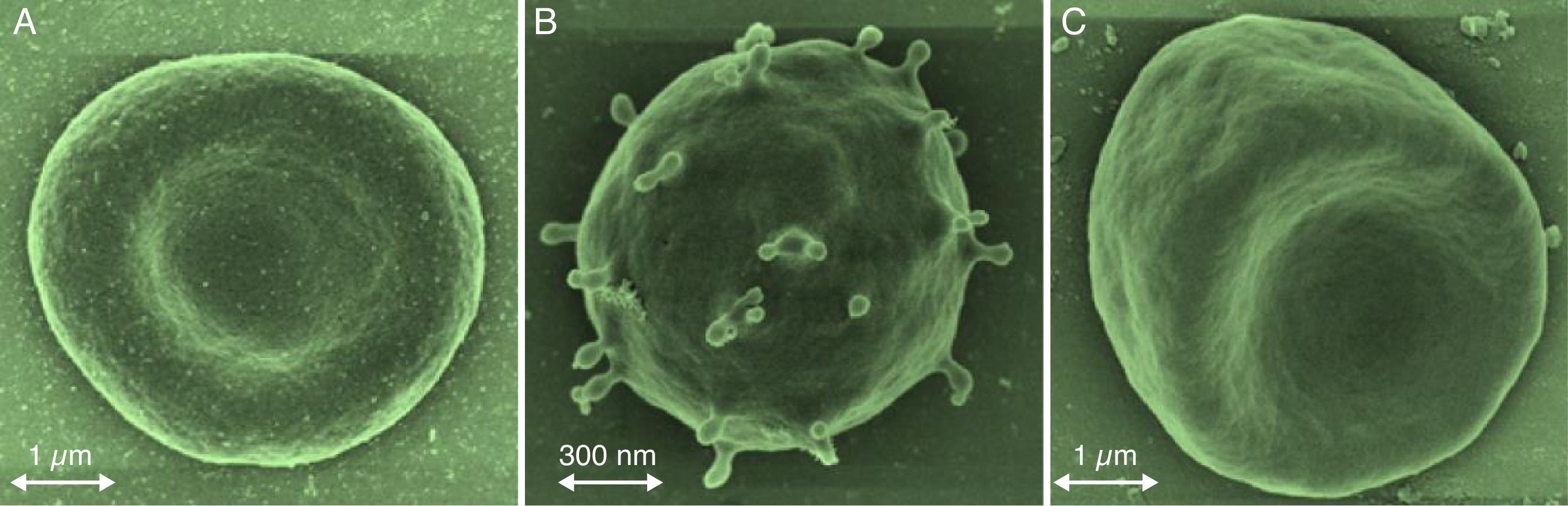
 Eritrocitos normales: forma bicóncava discoidal. B) Eritrocitos en forma de esferocitos con formación de microvesículas. C. Eritrocitos en forma de estomatocitos inducidos por estrés oxidativo. Las formas eriptóticas de esferocitos y equinocitos exponen fosfatidilserina y aumentan la probabilidad de adhesión al endotelio y la formación de trombos. Adaptada de Vittori y col. 2012.")
Cambios morfológicos en la eriptosis. A) Eritrocitos normales: forma bicóncava discoidal. B) Eritrocitos en forma de esferocitos con formación de microvesículas. C. Eritrocitos en forma de estomatocitos inducidos por estrés oxidativo. Las formas eriptóticas de esferocitos y equinocitos exponen fosfatidilserina y aumentan la probabilidad de adhesión al endotelio y la formación de trombos. Adaptada de Vittori y col. 2012.
Aunque la muerte programada de los eritrocitos ha sido planteada como un mecanismo de escape ante la hemólisis, otros estudios han dilucidado el papel de los eritrocitos eriptóticos en el deterioro de la microcirculación31,32. En la circulación, los glóbulos rojos están continuamente expuestos a fuentes endógenas y exógenas de especies reactivas de oxígeno que pueden generar daño estructural y alterar su función33. La eriptosis se activa como respuesta al estrés oxidativo o disminución de la defensa antioxidante12,23. Un estudio en pacientes diabéticos tipo 2 encontró una correlación positiva entre la lipoperoxidación inducida por el estrés y la externalización de la fosfatidilserina34.
Formación de la ceramida y la eriptosisLa ceramida es un importante lípido que actúa como segundo mensajero en muchos procesos celulares, incluyendo la apoptosis y la senescencia. Se obtiene de la hidrólisis de la esfingomielina a través de la acción de la enzima esfingomielinasa ácida, la cual origina la fosfocolina y la ceramida. Dicha enzima, no ha sido reportada como parte del proteoma de los eritrocitos, pero pueden estar expuestos a: la esfingomielinasa ácida secretada por el endotelio vascular, los leucocitos y las plaquetas. En el eritrocito, la ceramida dificulta las interacciones entre la membrana celular y el citoesqueleto, alterando la integralidad de la primera, favoreciendo de esta manera la exposición de la fosfatidilserina. La secreción de esta enzima puede estar aumentada en diversas situaciones incluyendo: la diabetes, la sepsis, las enfermedades cardiovasculares y las pulmonares, entre otras35,36.
La formación aumentada de la ceramida también se genera durante el estrés osmótico (fig. 1), en donde los eritrocitos activan la fosfolipasa A2 (PLA2), generando los lisoderivados que son transformados por el factor activador de plaquetas (PFA)35, el cual es un mediador de los fosfolípidos, involucrado en la regulación de: la inflamación, la trombosis, la aterogénesis y la función cardiovascular. En este mecanismo, se activa posiblemente una esfingomielinasa ácida que genera la ceramida por hidrólisis de la esfingomielina9,37. Estos procesos propician la vesiculización de la membrana celular estimulando la translocación de los fosfolípidos, desde su cara interna, produciendo la exposición de la fosfatidilserina, para luego ser reconocidos y degradados por los macrófagos que poseen receptores específicos para la fosfatidilserina. La acción de la PLA2, también genera el ácido araquidónico a partir de la fosfatidilcolina, dicho ácido es convertido a prostaglandina E2 por acción de la enzima ciclooxigenasa, la cual estimula el canal catiónico que permite la entrada de Ca2+ (fig. 1)24,35,36,38.
Adherencia de la fosfatidilserina a las células endotelialesNumerosos estudios indican que la inducción de la eriptosis conduce a un aumento significativo del número de eritrocitos firmemente adheridos a las células del endotelio vascular. Así mismo, las alteraciones de las proteínas de la membrana de los eritrocitos pueden propiciar enfermedades trombóticas1,2,16,19,39–41. Un estudio reciente encontró que las células eriptóticas que exponen la fosfatidilserina se adherían con facilidad al endotelio vascular a través de la proteína transmembrana CXCL1611. Ésta es una quimioquina expresada en las células endoteliales que ha sido implicada en el proceso aterosclerótico por participar como receptor de las lipoproteínas de baja densidad oxidadas (LDLox)42,43. Por tanto, las interacciones PS-CXCL16 aumentan la adhesividad de los eritrocitos, lo cual puede contribuir a la fisiopatología de las enfermedades cardiovasculares en los casos que exista eriptosis excesiva.
Se ha sugerido que el nivel circulante de CXCL16 podría ser un predictor de mortalidad a largo plazo en los síndromes coronarios agudos. Este receptor también es expresado en las células inflamatorias como las células dendríticas, los linfocitos T y los macrófagos cargados de lípidos en las placas ateroscleróticas44,45. La CXCL16 es fuertemente estimulada por las citoquinas proinflamatorias como el TNF-α e IFN-γ46, los cuales son mediadores asociados con enfermedades inflamatorias o isquemia11. Los eritrocitos que exponen la fosfatidilserina aumentan su adhesividad al endotelio en enfermedades asociadas con un mayor riesgo de complicaciones cardiovasculares, tales como: la diabetes o la insuficiencia renal crónica30,47,48.
Influencia de los fosfolípidos en las enfermedades cardiovascularesLa perturbación de la simetría de los fosfolípidos de la membrana de los eritrocitos puede inducir cambios morfológicos y externalización de la fosfatidilserina, y en última instancia esta puede conducir a la activación trombogénica de los eritrocitos2. Los fosfolípidos son una clase de lípidos complejos que se componen de: dos ácidos grasos, una unidad de glicerol, un grupo fosfato y una molécula polar49. En el eritrocito, por acción de la fosfolipasa D, la fosfatidilcolina genera el ácido fosfatídico50. El aumento de este último y de la actividad de la fosfolipasa D ha sido reportada en múltiples enfermedades como: la inflamación, la diabetes, la aterosclerosis, la hipertensión y los eventos trombóticos; lo que sugiere un mayor riesgo para las enfermedades cardiovasculares50,51. El ácido fosfatídico afecta: la proliferación celular, la reorganización del citoesqueleto y el tráfico vesicular, además, puede inducir actividades trombogénicas. Éste actúa a través de la activación de la PKC dependiente de Ca2+, mediando con ello la exposición de la fosfatidilserina con la consecuente eriptosis (fig. 3)30. Se ha sugerido que la activación de PKC puede influir en la integridad del citoesqueleto y las funciones de los eritrocitos, lo cual está mediado por la fosforilación de algunas proteínas de la membrana9.
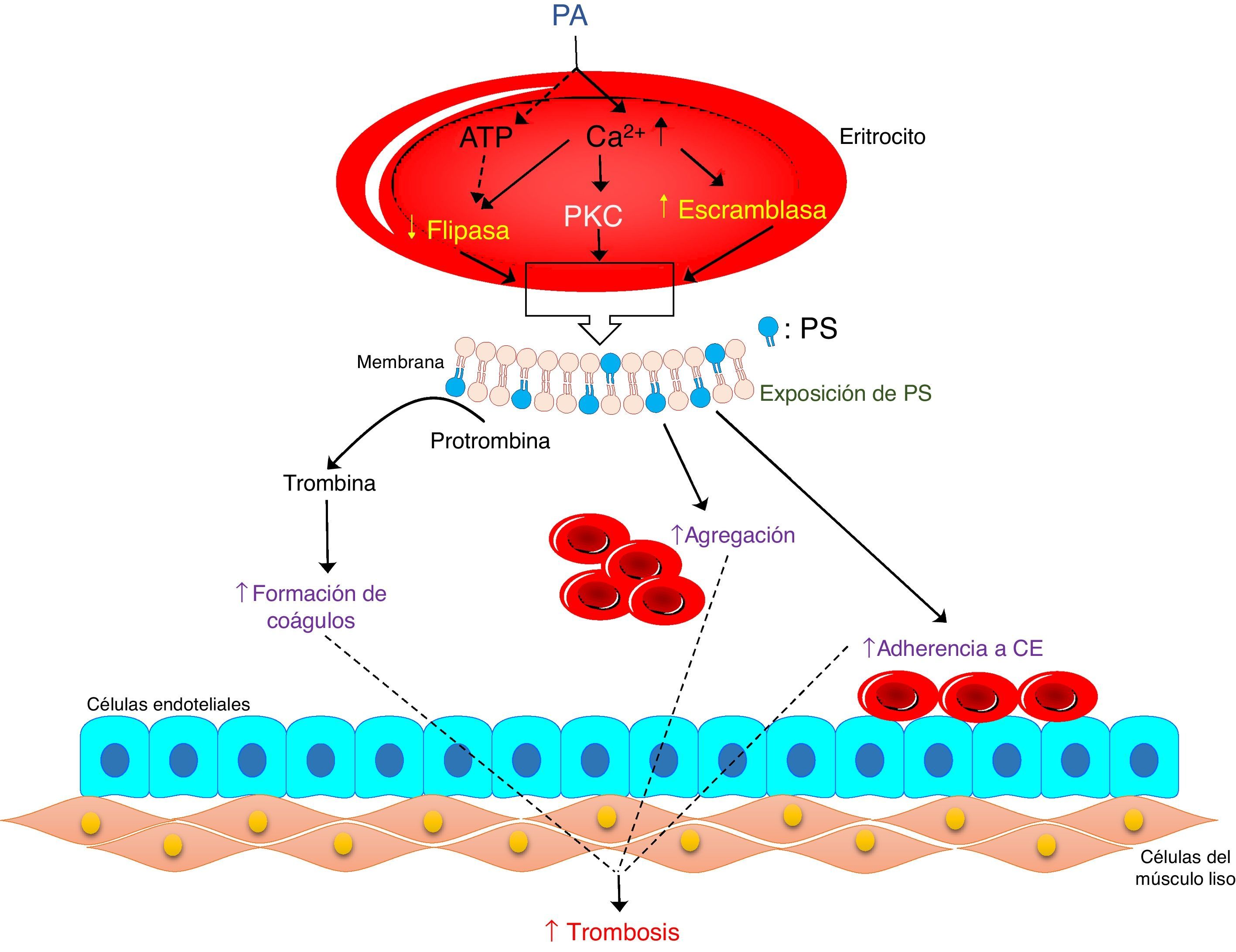
, lo cual está mediada por una disminución de ATP, aumento de Ca2+, y activación de PKC, que culmina con la activación de la escramblasa e inhibición de la flipasa. Los eritrocitos estimulados con PA pueden inducir la generación de trombina en el plasma y aumentar la agregación y adhesión de los eritrocitos a las células endoteliales, contribuyendo a la formación de trombos. PA: ácido fosfatídico. PS: fosfatidilserina. PKC: proteína quinasa C. CE: célula endotelial. Adaptado de Noh y col. 2010.")
Actividad trombogénica del ácido fosfatídico en eritrocitos humanos. Los eritrocitos responden a PA endógeno para expresar PS en su superficie (eriptosis), lo cual está mediada por una disminución de ATP, aumento de Ca2+, y activación de PKC, que culmina con la activación de la escramblasa e inhibición de la flipasa. Los eritrocitos estimulados con PA pueden inducir la generación de trombina en el plasma y aumentar la agregación y adhesión de los eritrocitos a las células endoteliales, contribuyendo a la formación de trombos. PA: ácido fosfatídico. PS: fosfatidilserina. PKC: proteína quinasa C. CE: célula endotelial. Adaptado de Noh y col. 2010.
La exposición de la fosfatidilserina proporciona un sitio de ensamblaje para las enzimas procoagulantes, con la consecuente generación de la trombina que facilita la hemostasia17,52. Otros lípidos endógenos como: el ácido araquidónico, las prostaglandinas y el factor activador de las plaquetas pueden inducir la exposición de este fosfolípido en los eritrocitos30.
En un modelo in vivo con ratas, la administración de ácido fosfatídico indujo aumento en la formación de trombos, con un efecto dependiente de la concentración del mismo30. Adicionalmente, el ácido fosfatídico puede originar el ácido lisofosfatídico por acción de la PLA2. Este último, también puede favorecer la eriptosis y actuar como una molécula aterogénica y trombogénica2,30. Generalmente, varios estudios han reportado que la relación entre la exposición de la fosfatidilserina y la microvesiculación están mediados por el aumento del calcio libre intracelular38. Sin embargo, otros autores han reportado que el ácido lisofosfatídico favorece la activación de PKC independiente del calcio. Otro aspecto relevante del ácido fosfatídico y el ácido lisofosfatídico, es el efecto inhibidor sobre la actividad de la flipasa, la cual es una proteína importante en el mantenimiento de la asimetría de la membrana2,30.
El ácido fosfatídico puede ser liberado por diversos estímulos, incluyendo: mediadores inflamatorios, hormonas y agonistas que participan en la activación de la fosfolipasa D en: los cardiomiocitos, las plaquetas, los neutrófilos y los eritrocitos47,53. Los eritrocitos que exponen la fosfatidilserina se vuelven más adhesivos a las células endoteliales siendo más propensos a la agregación eritrocitaria. Dichos eventos pueden contribuir a un mayor riesgo de desarrollar la formación de trombos, afectando con ello el sistema cardiovascular30.
Los metales pesados en la enfermedad cardiovascularAunque pocos estudios han centrado su atención en los efectos tóxicos de los metales pesados en el sistema cardiovascular, estudios recientes demuestran la asociación del mercurio con: la hipertensión, la aterosclerosis, el infarto del miocardio y la enfermedad arterial coronaria54–58. Las teorías bioquímicas más aceptadas hasta la fecha indican una posible inducción del proceso de muerte celular de los eritrocitos por el estrés oxidativo, los cuales exponen la fosfatidilserina. Algunos estudios experimentales in vivo (ratas) evidenciaron que la exposición al mercurio induce la disfunción endotelial en las arterias coronarias54,55,59.
Los efectos del mercurio en el sistema cardiovascular parecen ser dependientes tanto de la dosis como del tiempo de exposición. Estudios en humanos han reportado que la exposición crónica al mercurio puede aumentar la resistencia vascular e inducir la hipertensión56. Por tanto, la presencia de este metal en el organismo puede ser considerado un factor de riesgo para las enfermedades cardiovasculares. La gravedad de la exposición al mercurio se debe a que tiene alta afinidad por grupos tioles o sulfhidrilos de las proteínas, lo que conlleva a la inactivación de numerosas reacciones enzimáticas, aminoácidos y antioxidantes, con la subsiguiente disminución de la defensa antioxidante y aumento del estrés oxidativo57.
Por otra parte, investigaciones in vitro concluyen que la exposición a dosis bajas de Hg2+ (0,25 - 5μM) conducen a cambios en la morfología de los eritrocitos28,55,58, acompañado de la generación de microvesículas y la exposición de la fosfatidilserina. Las interacciones del Hg2+ con las proteínas pueden ser un mediador para el aumento del Ca2+ intracelular libre y disminución del ATP, con la consecuente apoptosis del eritrocito28. Hasta la fecha el mecanismo de inducción de translocación de la fosfatidilserina por parte del mercurio no está claro. Una hipótesis sugiere la unión de estos iones metálicos a hPLSCR1 (escramblasa de fosfolípidos humana 1), lo cual favorece la exposición de la fosfatidilserina59. La exposición de este fosfolípido puede servir como un sitio para el anclaje de un complejo protrombinasa y tenasa, dando lugar a la generación de trombina con cambios en la forma del eritrocito que pueden fomentar el aumento de la adherencia de estas células a la pared vascular y la formación de trombos28. Todas estas evidencias muestran cada vez una mayor relación del mercurio con las enfermedades cardiovasculares. No obstante, se necesitan más estudios para comprender totalmente los mecanismos implicados en los efectos del mercurio en el sistema cardiovascular.
Inhibidores de la eriptosisLa inhibición de los canales catiónicos del calcio por algunas sutancias como: el amiloride y el etilisopropilamiloride60, han mostrado una prevención del proceso de muerte celular del eritrocito. En otros estudios, la dopamina, el isoproterenol y la epinefrina han mostrado similares efectos61. Lo anterior se explica porque el aumento del Ca2+ citosólico y la pérdida celular del K+ participan en la activación de la escramblasa de los eritrocitos y por consiguiente el proceso de eriptosis. Por otro lado, otros estudios reportan que el clotrimazol o la caribdotoxina aumentan los niveles del K+ contrarrestando la pérdida de este catión e inhibiendo el proceso de eriptosis62.
Otras enfermedades asociadas a la eriptosisLa eriptosis se ha relacionado con una variedad de condiciones clínicas o enfermedades, incluyendo: el síndrome urémico hemolítico63, la sepsis64, la malaria65, las células falciformes, la β talasemia, la deficiencia de glucosa-6-fosfato deshidrogenasa, la esferocitosis hereditaria, la hemoglobinuria paroxística nocturna, el síndrome mielodisplásico, el agotamiento de fosfato, la deficiencia de hierro, la insuficiencia renal, la insuficiencia cardíaca, etc.
Síndrome urémico hemolíticoEl síndrome urémico hemolítico (SUH), se caracteriza por la anemia hemolítica con presencia de fragmentados eritrocitarios, trombocitopenia y falla renal aguda63. Frecuentemente, el SUH es causado por las verotoxinas producidas por Escherichia coli O157:H766, y de forma infrecuente, por un defecto genético de las proteínas del complemento o las proteasas asociadas al factor de Von Willebrand67,68. El SUH puede también ser causado por ciertos fármacos incluyendo: la ciclosporina, los anticonceptivos orales, la quinina y la cocaína69. Los estudios realizados por Lang, et al. 63, muestran que el plasma de pacientes con SUH provoca la muerte suicida de los eritrocitos, demostrando con ello que la exposición de eritrocitos a plasma de pacientes con SUH produce: el ingreso del Ca2+, la formación de ceramida y la exposición de PS. Lo anterior favorece la unión a receptores de PS expresados por los macrófagos y su fagocitosis, eliminando de esta manera a los eritrocitos afectados.
SepsisEs una condición clínica asociada a infecciones con una amplia variedad de patógenos. Entre las secuelas características de la sepsis se puede nombrar el desarrollo de la anemia; no por una disminución en la formación de los eritrocitos sino por una eliminación acelerada de los mismos de la circulación70. Cuando los eritrocitos de voluntarios sanos se exponen al plasma de pacientes sépticos o sobrenadantes de patógenos, se incrementa la exposición de PS. El efecto del plasma de pacientes sépticos sobre la concentración del Ca2+ es moderado como para ser responsable de la fuerte estimulación de la exposición de PS. En este caso, es probable que la formación de la ceramida por ruptura de SM mediada por la esfingomielinasa sea el factor más importante. Varias bacterias patógenas producen las esfingomielinasas que estimulan la formación de la ceramida. La hemólisis inducida por determinados patógenos puede de manera similar contribuir a la eliminación acelerada de los eritrocitos y producir la anemia en pacientes sépticos, como se ha demostrado luego de la infección por Clostridium perfringens64.
La malariaEl patógeno causante de la malaria es el Plasmodium falciparum, que entra en el eritrocito escapando del reconocimiento por el sistema inmune del huésped. Es por eso que el Plasmodium falciparum depende de la activación de canales iónicos en el eritrocito que le permiten incorporar nutrientes, Na+ y Ca+2. A su vez, activa los canales en el eritrocito por el estrés oxidativo produciendo la apertura de los canales catiónicos permeables al Ca+2 que estimulan la eriptosis. Mientras que los canales se requieren para la sobrevida, la replicación y la maduración del Plasmodium falciparum en el eritrocito, también limitan la vida del eritrocito infectado y en consecuencia del patógeno intracelular65.
Deficiencia de hierroLa deficiencia de hierro ocasiona la anemia, la cual en parte es debida a una disminución de la vida media del eritrocito. Está aceptado que la anemia por deficiencia de hierro es el resultado de una eritropoyesis disminuida71. Sin embargo, existen algunas evidencias que indican que la anemia en este caso también es ocasionada por una vida media disminuida de los eritrocitos deficientes en hierro. El contenido reducido de hemoglobina en eritrocitos deficientes en hierro lleva a una disminución de la presión osmótica coloidal, y por tanto, a una reducción del volumen celular. Esta disminución de volumen es responsable de un aumento en la actividad de los canales catiónicos no selectivos. Aunque el ingreso del Na+ a través de este canal produce un incremento de volumen, la entrada del Ca+2 ejerce un efecto opuesto. El ingreso del Ca+2 y la activación del canal Gardos contribuyen a la activación de la eriptosis. Además, dado que los eritrocitos deficientes en hierro son más sensibles al estrés oxidativo, esto induce la activación de los canales catiónicos24. Esta condición puede agravarse en pacientes con insuficiencia cardiaca o insuficiencia renal por incremento de la eriptosis32,72.
Por otra parte, los nuevos mecanismos observados de la eriptosis presumen que pueden tener una relación intrínseca con la producción de enfermedades cardiovasculares como: la trombosis, los infartos, la ateroesclerosis o la hipertensión; dado que los eritrocitos que exponen la fosfatidilserina (PS), se vuelven más adhesivos a las células endoteliales, siendo más propensos al proceso de agregación eritrocitaria dando origen a las enfermedades cardiovasculares30.
ConclusionesLas investigaciones en este campo de estudio señalan que el eritrocito posee una maquinaria bioquímica compleja, permitiendo la autodestrucción programada de los eritrocitos lesionados en circulación antes de la senectud.
La muerte del eritrocito consta de varias vías de señalización que incluyen: la formación de la ceramida, el aumento del calcio intracelular y la activación de la escramblasa. Cabe resaltar que el punto clave de la eriptosis es la exposición a la fosfatidilserina, que posee receptores de reconocimiento por parte de los macrófagos para su eliminación. La eriptosis acelerada ha sido blanco para numerosos estudios que han relacionado la exposición de la fosfatidilserina como un vínculo molecular para la adhesión a la pared vascular, con la consecuente formación de trombos. El conocimiento de los nuevos mecanismos que subyacen a la formación de trombos puede ser importante para la prevención clínica de las enfermedades cardiovasculares. Sin embargo, es necesario llevar a cabo otros estudios que busquen esclarecer la señalización de este proceso y sus implicaciones en la actividad procoagulante por parte de los eritrocitos eriptóticos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener conflictos de intereses.
A la Universidad de Cartagena especialmente a la Vicerrectoría de Investigaciones en el plan de fortalecimientos de grupos de investigación.