



La PCSK9 es una proteína que se une a los receptores de LDL, precipita la degradación de estos últimos y por tanto eleva los niveles plasmáticos de colesterol LDL (cLDL). La inhibición de la PCSK9 con anticuerpos monoclonales evita la degradación de los receptores, permite su reciclaje y la mayor captación de cLDL y reduce eventos cardiovasculares.
Los anticuerpos monoclonales contra PCSK9, evolocumab y alirocumab, son una novedosa herramienta terapéutica de gran utilidad en los casos de hipercolesterolemia familiar heterocigota, en algunos casos de hipercolesterolemia familiar homocigota y de personas con alto riesgo cardiovascular, cuyos niveles de cLDL no pueden ser controlados con dosis máximas toleradas de las estatinas de mayor efectividad, además de ezetimibe y cambios en el estilo de vida.
PCSK9 is a protein which binds to LDL receptors and precipitates their breakdown, thereby elevating plasma LDL cholesterol (LDL-C). Inhibition of PCSK9 with monoclonal antibodies avoids the breakdown of the receptors, allows them to be recycled, leads to a greater capture of LDL-C, and reduces cardiovascular events.
Evolocumab and alirocumab, monoclonal antibodies against PCSK9, are very useful novel therapeutic tools in heterozygous familial hypercholesterolemia, some homozygous familial hypercholesterolemia cases, and in individuals with a high cardiovascular risk whose LDL-C levels cannot be controlled with maximum tolerated doses of highly effective statins, ezetimibe, and lifestyle changes.
La aterosclerosis y sus manifestaciones principales son la primera causa global de muerte y pronto también la primera causa de morbilidad. Los esfuerzos para su control incluyen medicamentos de efectividad y seguridad probada, cambios terapéuticos en estilo de vida y campañas nacionales e internacionales para el logro de metas y control de los factores de riesgo.
Sin embargo, aún queda mucho riesgo residual sin controlar, y la investigación no se detiene en cuanto a la búsqueda de nuevas opciones. Entre las causas para el control insuficiente se han demostrado barreras en el acceso a la atención en salud, falta de adherencia a los cambios en estilo de vida y a los medicamentos, y suspensión frecuente de las estatinas, por intolerancia real en ciertos casos1,2.
Al tratamiento de las hipercolesterolemias se agregó en la última década una nueva intervención, una familia de medicamentos particularmente efectivos en la reducción de los niveles de cLDL, que son también seguros y que tienen un esquema práctico de aplicación, que consiste en una inyección subcutánea quincenal o mensual. Se trata de un biológico formado por anticuerpos monoclonales específicos contra la PCSK9, una proteína cuya función es inactivar los receptores para LDL1 en los hepatocitos. Al inhibir esa proteína se protegen los receptores, que pueden entonces no sólo cumplir su función de captura de LDL, sino recircular2 (cada ciclo dura entre 10 y 20 minutos) varios cientos de veces durante sus 20 horas de vida3.
Proproteína convertasa de subtilisina kexina 9 (PCSK9): una nueva opción terapéuticaGoldstein y Brown cambiaron la historia de la enfermedad cardiovascular al anunciar su descubrimiento del receptor para LDL, que significó un enorme avance en la comprensión de la biología celular, porque no sólo se demostró el mecanismo de endocitosis mediada por un receptor4 sino que se estableció que los receptores podían ser reciclados después de cumplir su función, y finalmente, que la expresión y sobrevida de los receptores estaba cuidadosamente regulada por retroalimentación.
Más recientemente hubo un nuevo descubrimiento, que completó la información sobre los receptores de LDL. El grupo de Nabil Seidah en Montreal5 identificó una mutación en la que la ganancia en función de un gen era la responsable de hipercolesterolemia familiar (HF) en una familia. Poco después se encontró6 que la pérdida de función del mismo gen estaba asociada tanto con valores muy bajos de cLDL como con incidencia muy reducida de enfermedad cardiovascular7. Esto llevó al descubrimiento del papel de una proproteína, la PCSK9, y después al desarrollo de anticuerpos monoclonales para inhibirla8, con el resultado de un biológico que logra reducciones significativas en el colesterol LDL11,12 y también en eventos cardiovasculares9.
Control de los niveles séricos de colesterolEl colesterol, sintetizado principalmente en el hígado, está presente en las células animales y es una fuente importante de energía. Es también vital para la producción de algunas hormonas fundamentales para el desarrollo y para la expresión de características sexuales, así como para el balance de sodio y agua (hormonas mineralocorticoides), la respuesta inflamatoria, la inmunidad, la formación de algunas vitaminas y la estructura de las membranas celulares14,15.
El colesterol es producido principalmente por el hepatocito. En este sentido, el trabajo de Goldstein y Brown fue el pilar fundamental para la comprensión de los mecanismos de regulación cuando decidieron medir la actividad de la reductasa de HMG-CoA, una enzima que regula un paso limitante en la génesis de colesterol, en extractos de cultivos de fibroblastos10,11. Notaron que cuando se cultivaban fibroblastos con suero humano normal, la actividad de la enzima era baja. Sin embargo, cuando se quitaban las lipoproteínas transportadoras de colesterol del medio de cultivo, la actividad de la enzima aumentaba hasta cincuenta veces en 24 horas, pero si se agregaba LDL al medio, se suprimía rápidamente la actividad enzimática inducida. Esto los llevó a postular la presencia de un receptor muy específico para LDL, la única lipoproteína capaz de lograr los cambios enzimáticos4.
La historia se desarrolló rápidamente y el descubrimiento de los receptores tuvo aplicación en muchos aspectos, uno de ellos el desarrollo de las estatinas12, medicamentos con efectividad probada para la reducción del cLDL y de eventos cardiovasculares13. Las lipoproteínas son transportadores de triglicéridos y de colesterol y además de su capa externa de fosfolípidos, que les permite solubilidad en el plasma, tienen unas proteínas externas, las apoproteínas, que les sirven como ligandos para los receptores específicos14. El control de niveles de colesterol depende principalmente de la síntesis, que a su vez está determinada como paso limitante por la expresión de la reductasa de HMG-CoA y por la captación de LDL en la superficie celular.
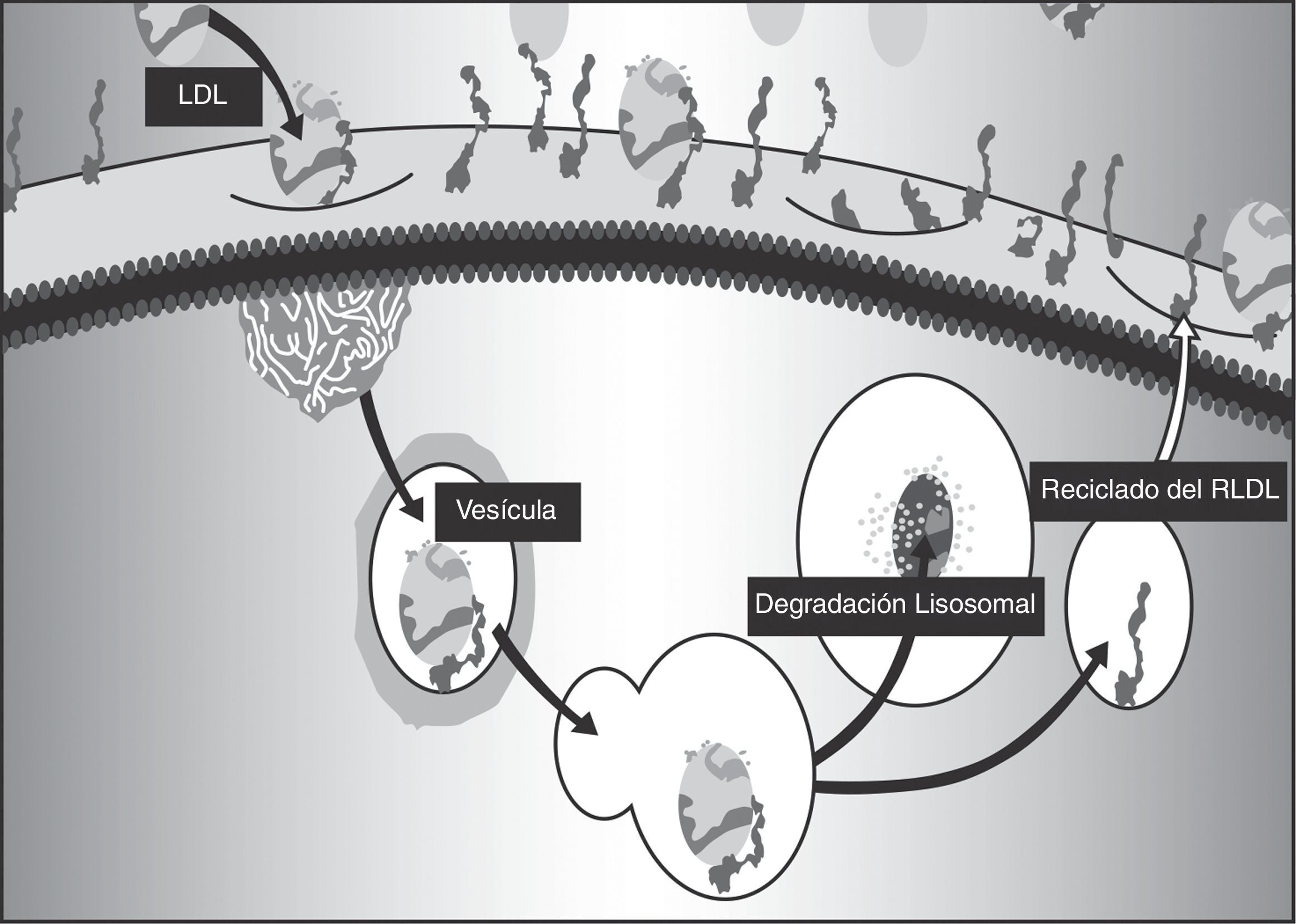
Papel de los receptores para LDLEn la regulación de la síntesis y utilización del colesterol los receptores desempeñan un papel crucial, puesto que captan el colesterol transportado en las LDL, que ingresa a la célula por un mecanismo de endocitosis. La unión al receptor necesita un receptor funcional, una proteína adaptadora del receptor de LDL (LDLRAP1) y la apoproteína B100 de la LDL15. Una vez unida al receptor, la LDL es internalizada (fig. 1).

Los receptores también están regulados por la hormona tiroidea y pueden estar sobrerregulados en el hipertiroidismo, lo que se acompaña de valores bajos de lípidos, o subregulados en el hipotiroidismo, fenómeno que explica la hipercolesterolemia secundaria que lo acompaña16.
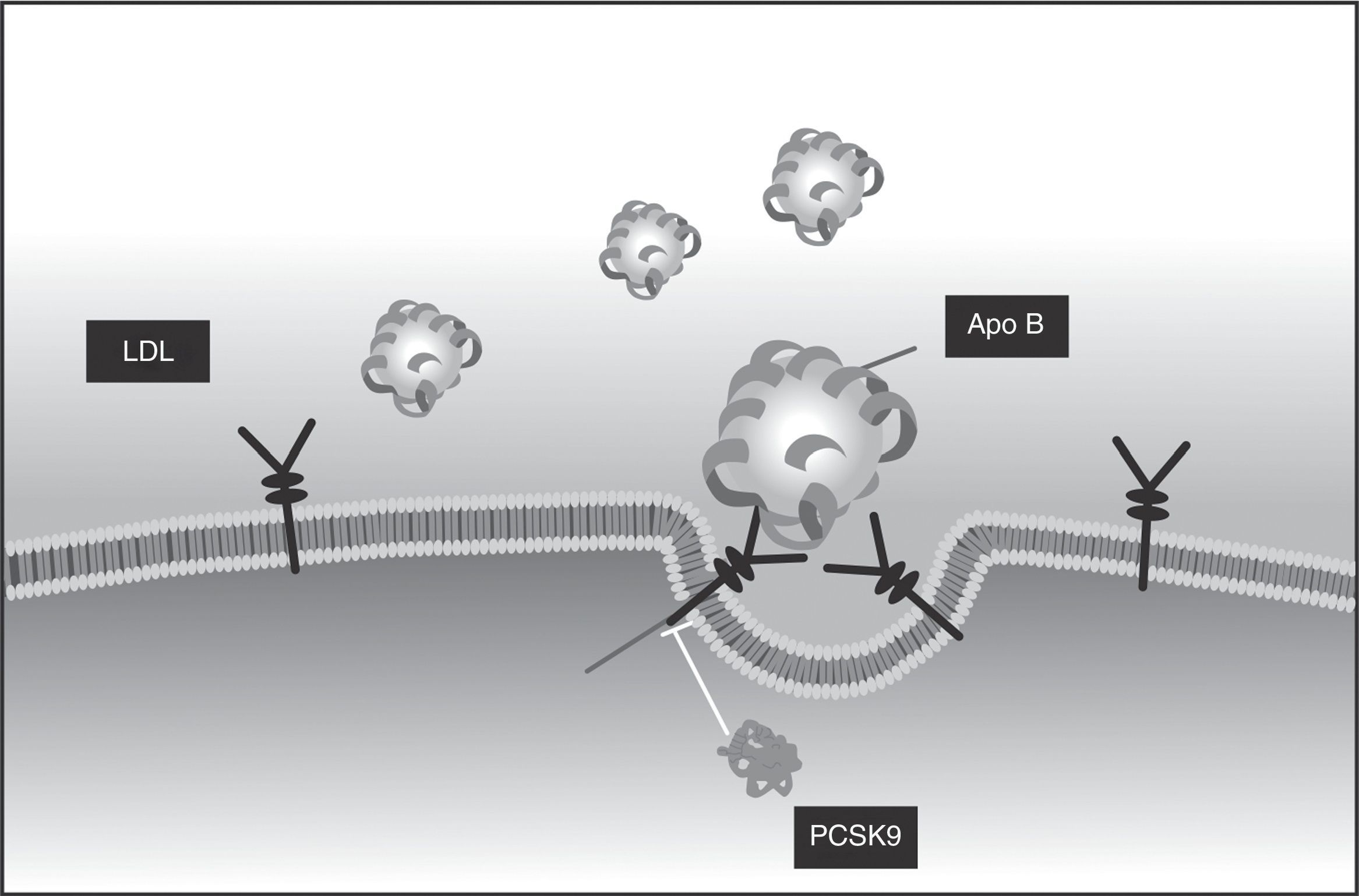
PCSK9Las convertasas de proproteínas tienen como función la conversión a la forma activa de proteínas secretorias, como hormonas, receptores o aún enzimas. Una de ellas, la PCSK9, es codificada por el gen PCSK9 y se expresa principalmente en el hígado. Su función es la regulación de los receptores de superficie para LDL, a los que se une en el exterior del hepatocito. Una vez adentro, se produce el clivaje y la lipoproteína es degradada para la utilización de sus componentes, y el receptor, que debería volver a la superficie para cumplir su función, no lo puede hacer porque la PCSK9 facilita su destrucción por los lisosomas17.
La PCSK9 se une al receptor de LDL en la superficie celular y solamente se separa cuando el complejo LDL-PCSK9 haya sido internalizado18. Sin embargo, por el efecto del medio ácido del endosoma19, la afinidad de la PCSK9 por una estructura específica del receptor puede aumentarse hasta 150 veces. Se ha probado que dicha estructura, similar al factor de crecimiento epidérmico (EGF-like), es fundamental para que el receptor de LDL vuelva a salir a la superficie celular20, y la unión cambia el itinerario del receptor y facilita su degradación por los lisosomas21; como el resultado el reciclaje de receptores se ve alterado y la disponibilidad de estos en la superficie celular se ve claramente disminuida22.
La actividad aumentada de PCSK9 trae como consecuencia la disminución en el número de receptores para LDL, además de aumento de niveles extracelulares de colesterol, lo cual, a su vez, se acompaña de incremento en la expresión de reductasa de HMG-CoA por la menor disponibilidad intracelular. Estos dos mecanismos, captación disminuida y producción aumentada, son los responsables de la hipercolesterolemia en los casos en los que hay menor cantidad o función de receptores para LDL.
La síntesis de RLDL depende de las concentraciones intracelulares de colesterol libre, que pueden, cuando están aumentadas, inhibir la transcripción del gen del receptor. No obstante, también puede controlarse la población de RLDL mediante la PCSK9, que se produce en mayor cantidad cuando hay incremento del colesterol intracelular. Una vez fuera de la célula se une al complejo de LDL/RLDL e induce la degradación del receptor cuando está nuevamente en el interior de ésta23.
Estos mismos mecanismos pueden utilizarse de manera inversa en el tratamiento de la hipercolesterolemia: las estatinas disminuyen la actividad de la reductasa al bloquearla, por lo cual se reduce la producción de colesterol, y, de manera secundaria, incrementan la expresión de receptores para LDL.
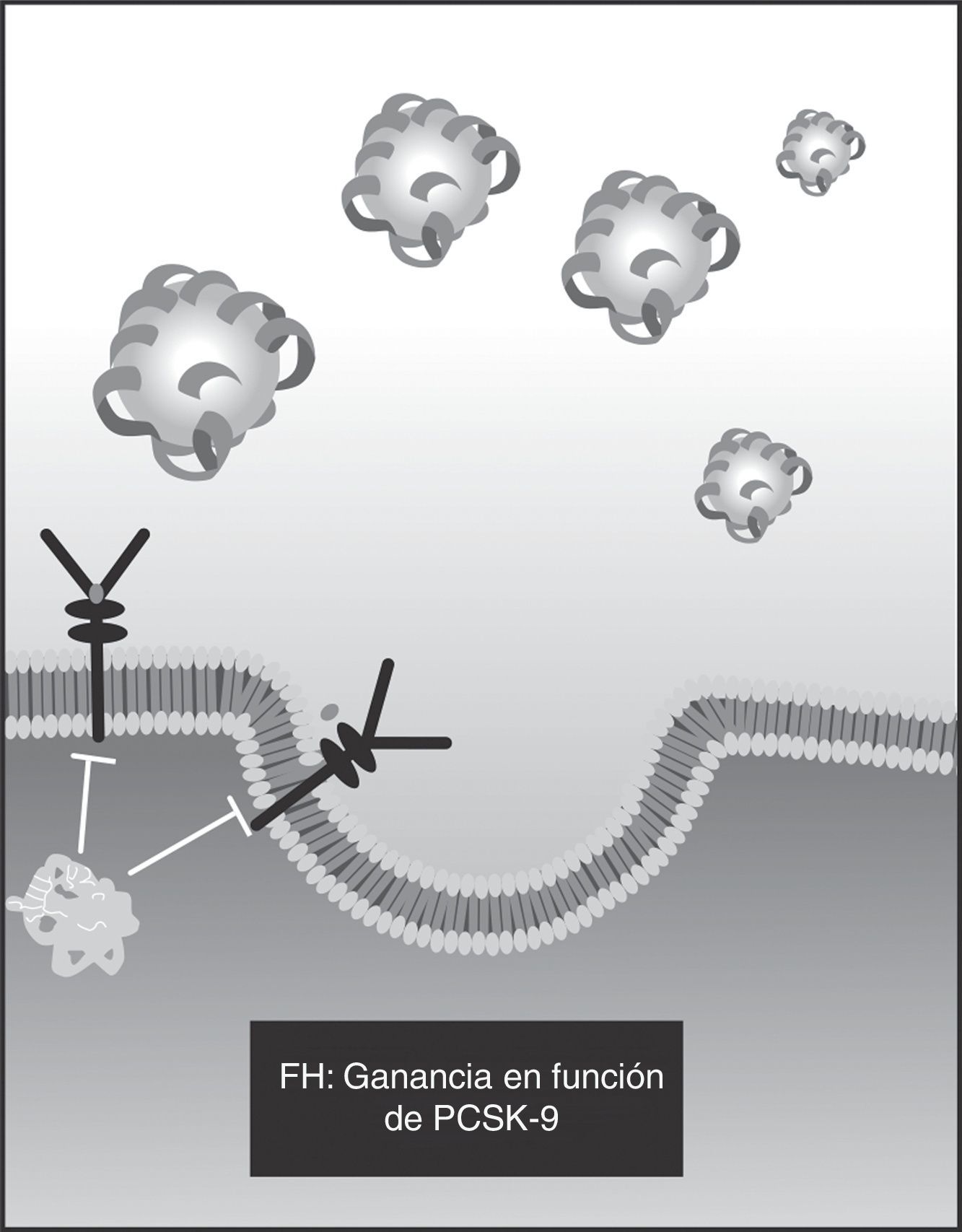
Este mecanismo es importante dado que explica la utilidad de las estatinas en la mayoría de casos de hipercolesterolemia, pero también su falta de efectividad en los casos de hipercolesterolemia familiar homocigota (HF Ho), en la cual no hay receptores (lo menos frecuente) o son claramente defectuosos (hasta en 90% de los casos). Esto explica por qué en la ganancia de función del gen PCSK9 se produce hipercolesterolemia, que se acompaña de aumento significativo en la enfermedad aterosclerótica: la mayor función del PCSK9 disminuye la cantidad de receptores disponibles24.
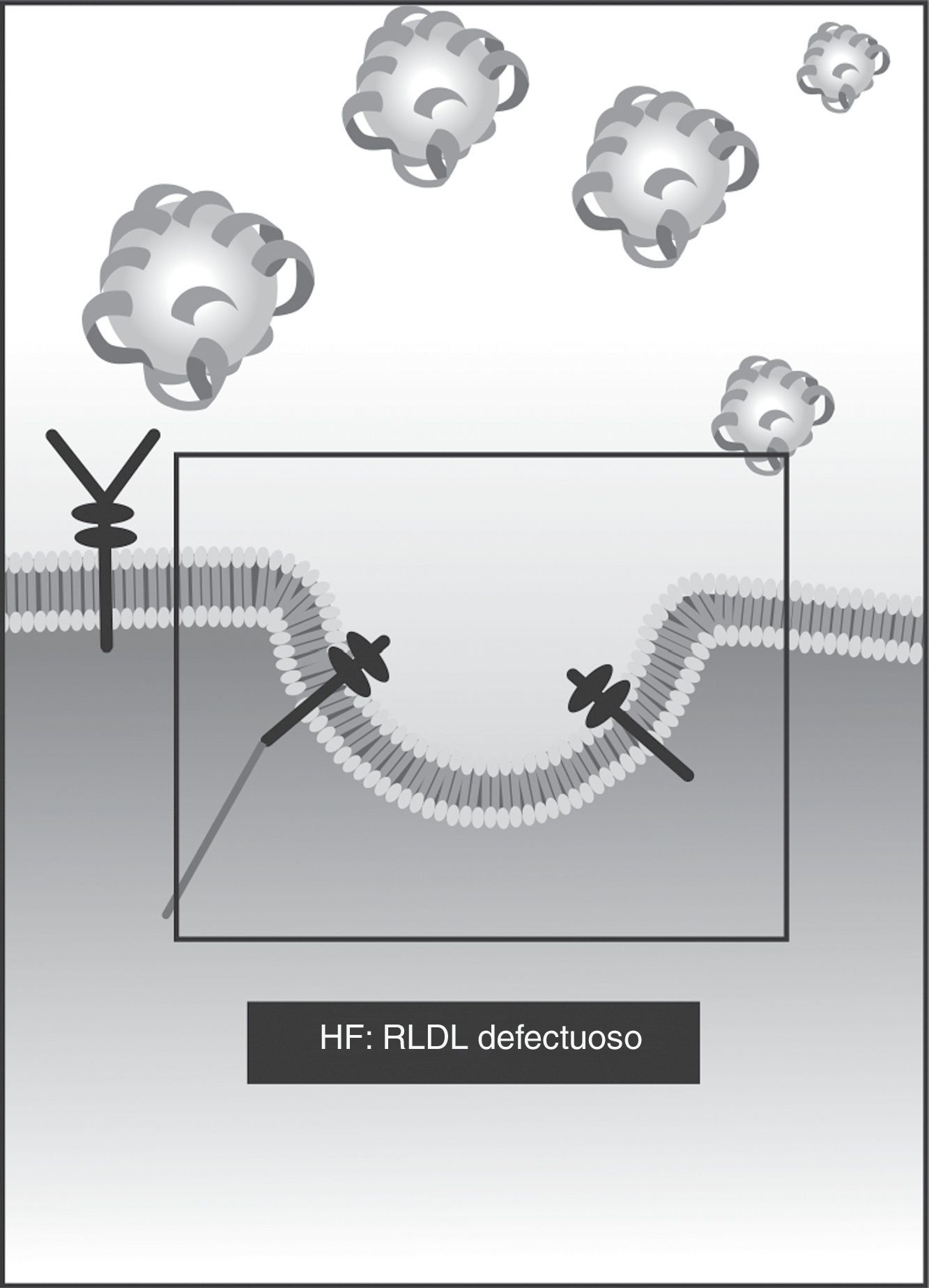
Mecanismos fisiopatológicos en la hipercolesterolemia familiarLas características principales de la hipercolesterolemia familiar (HF) son la elevación marcada de los niveles de LDL y la enfermedad aterosclerótica (principalmente coronaria) prematura. Las causas de la HF son diversas alteraciones genéticas (tabla 1) en la captación de LDL por parte de los receptores de superficie en el hepatocito (fig. 1). La más frecuente de las alteraciones es el defecto en la cantidad o calidad de los RLDL25 (> 90% de los casos), una mutación autosómica dominante. La alteración puede darse por ausencia de receptores (< 2% de la población de receptores), denominada actividad nula, o por actividad defectuosa (entre 2% y 25%) (fig. 2).
Principales causas genéticas de hipercolesterolemia familiar
| Gen comprometido | Herencia | Alteración |
|---|---|---|
| LDLR | Autosómica dominante | Reducción en número o función de receptores para LDL |
| APOB | Autosómica dominante | Alteración en la función de la apoB, que impide la unión de la LDL al receptor |
| PCSK9 | Autosómica dominante | Destrucción aumentada de los receptores para LDL |
| LDLRAP1 | Autosómica recesiva | Alteración en el adaptador del receptor, que dificulta la unión con la LDL |

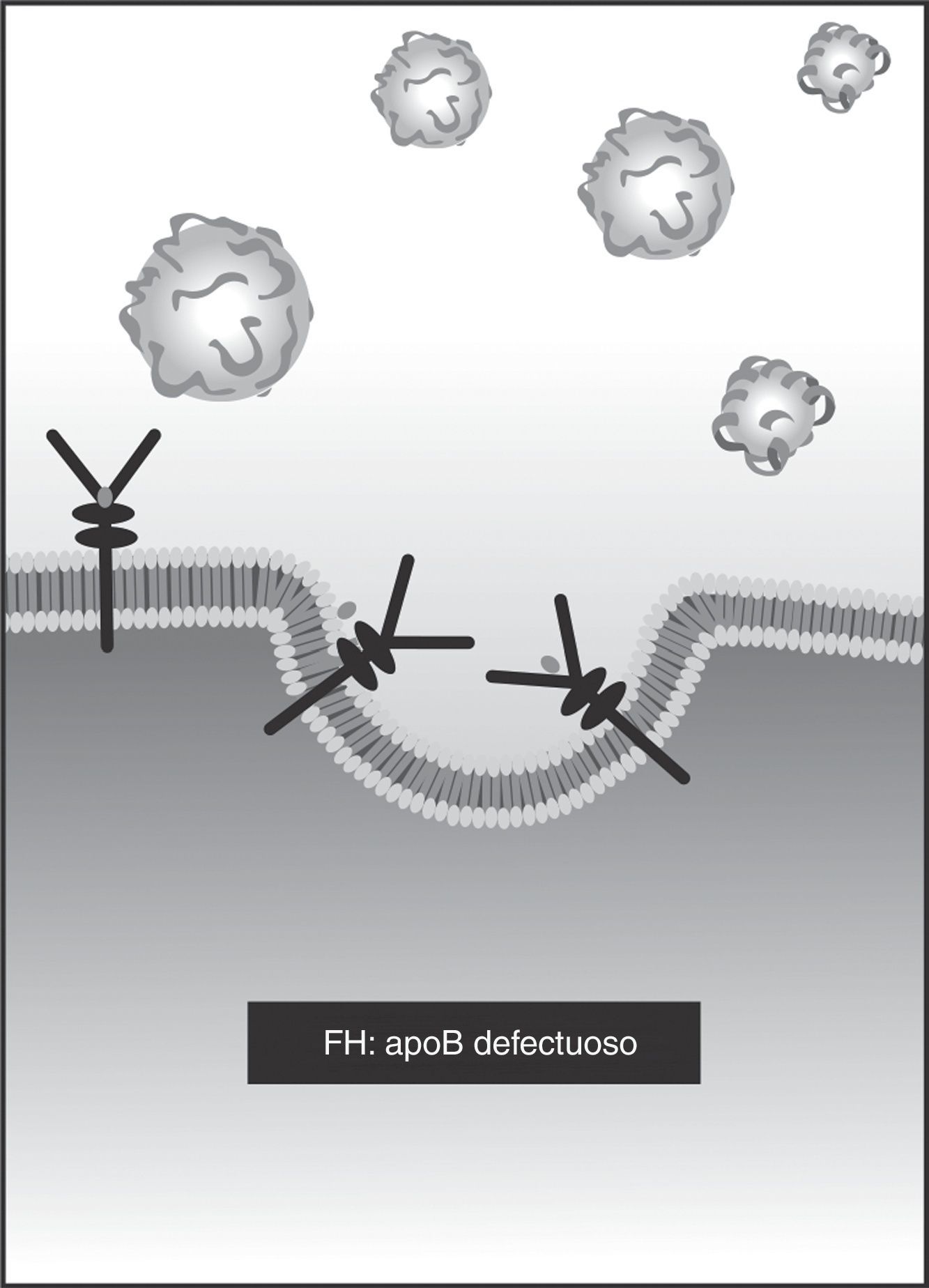
También puede haber defectos en el gen APOB, que codifica la proteína apoB26 (3-4% de los casos). Es un defecto autosómico dominante que impide la unión de la LDL al receptor, con el mismo resultado de acumulación extracelular de colesterol y aumento de la síntesis celular (fig. 3).

Un tercer defecto, mediante el cual se descubrió la PCSK9, es precisamente aquel en el gen PCSK9, que codifica la proproteína y es también autosómico dominante (presente en 1% de los casos) (fig. 4).

Finalmente, el cuarto mecanismo es el único trastorno autosómico recesivo, el defecto en el gen LDLRAP1, que codifica el adaptador para el receptor de LDL (presente en<1% de los casos de HF).
Importancia del mecanismo fisiopatológicoDe la causa del trastorno principal en la FH depende la respuesta a los medicamentos que se utilicen. Las estatinas, que disminuyen la expresión de reductasa de HMG-CoA y secundariamente afectan la expresión de los receptores para LDL, pueden tener efectividad en la hipercolesterolemia secundaria y en la primaria poligénica27. Su utilidad está limitada parcialmente en la HF heterocigota, en la que su efectividad depende de los niveles basales de LDL. En la HF homocigota, con las dos copias defectuosas, la efectividad es muy baja e incluso nula. Lo mismo puede ocurrir con los anticuerpos monoclonales contra la PCSK9. Si el trastorno genético implica actividad nula, la respuesta también lo será, pues si no hay producción de receptores no puede haber preservación de estos.
Sin embargo, en los casos en los que la expresión de receptores está disminuida (que es la mayoría), o cuando el mecanismo de hipercolesterolemia no los involucra, la efectividad puede ser muy alta28.
Anticuerpos monoclonales contra PCSK-9 (alirocumab y evolocumab)La observación de la baja frecuencia de enfermedad en pacientes con ganancia de función del gen PCSK9 llevó a la investigación para el desarrollo de nuevas opciones terapéuticas, en busca de que se disminuyera la función de la PCSK9 y se aumentara la disponibilidad de receptores para LDL29. Se estudiaron inicialmente las posibilidades de disminuir los niveles de PCSK9 o de inhibirla30. Para inhibir o alterar la unión de la PCSK9 al rLDL se consideraron opciones como el desarrollo de anticuerpos monoclonales, que dificultaran la unión al rLDL, o la producción de proteínas de unión como adnectinas, o el desarrollo de pequeñas moléculas inhibidoras31. Esta última opción resultó particularmente difícil porque la superficie de la PCSK9 no tiene espacio para la unión de otras moléculas32.
El proceso de desarrollo de anticuerpos monoclonales había tenido auge en los últimos años; había varios ejemplos en uso en la medicina y se decidió iniciar esta estrategia. La experiencia desde los años setenta ha mostrado que los anticuerpos monoclonales tienen buen perfil de seguridad y de efectividad a largo plazo, en inyecciones repetidas, con baja incidencia de reacciones inmunes del huésped, poca hipersensibilidad y prácticamente efectos nulos de citotoxicidad39–41, especialmente en los anticuerpos humanos, que ofrecen múltiples ventajas sobre los anticuerpos humanizados o sobre los quiméricos, que contienen fragmentos murinos.
Con el objetivo de crear los anticuerpos monoclonales se desarrollaron tres moléculas que se unen directamente al blanco terapéutico, la PCSK9. Dos de las moléculas, ya estudiadas y aprobadas por la FDA y por la EMA para uso clínico (así como por el INVIMA en Colombia), evolocumab y alirocumab, son humanas, en tanto que la tercera, bococizumab, cuyo programa de investigación fue suspendido, es humanizada. Es posible que sea esta justamente la razón (que no fuera completamente humana), ya que hubo casos de reacciones adversas en el sitio de inyección y de reducción de efectividad, secundaria a la generación de anticuerpos neutralizantes, así como una importante variabilidad en el efecto reductor de cLDL, aún en ausencia de receptores neutralizantes33.
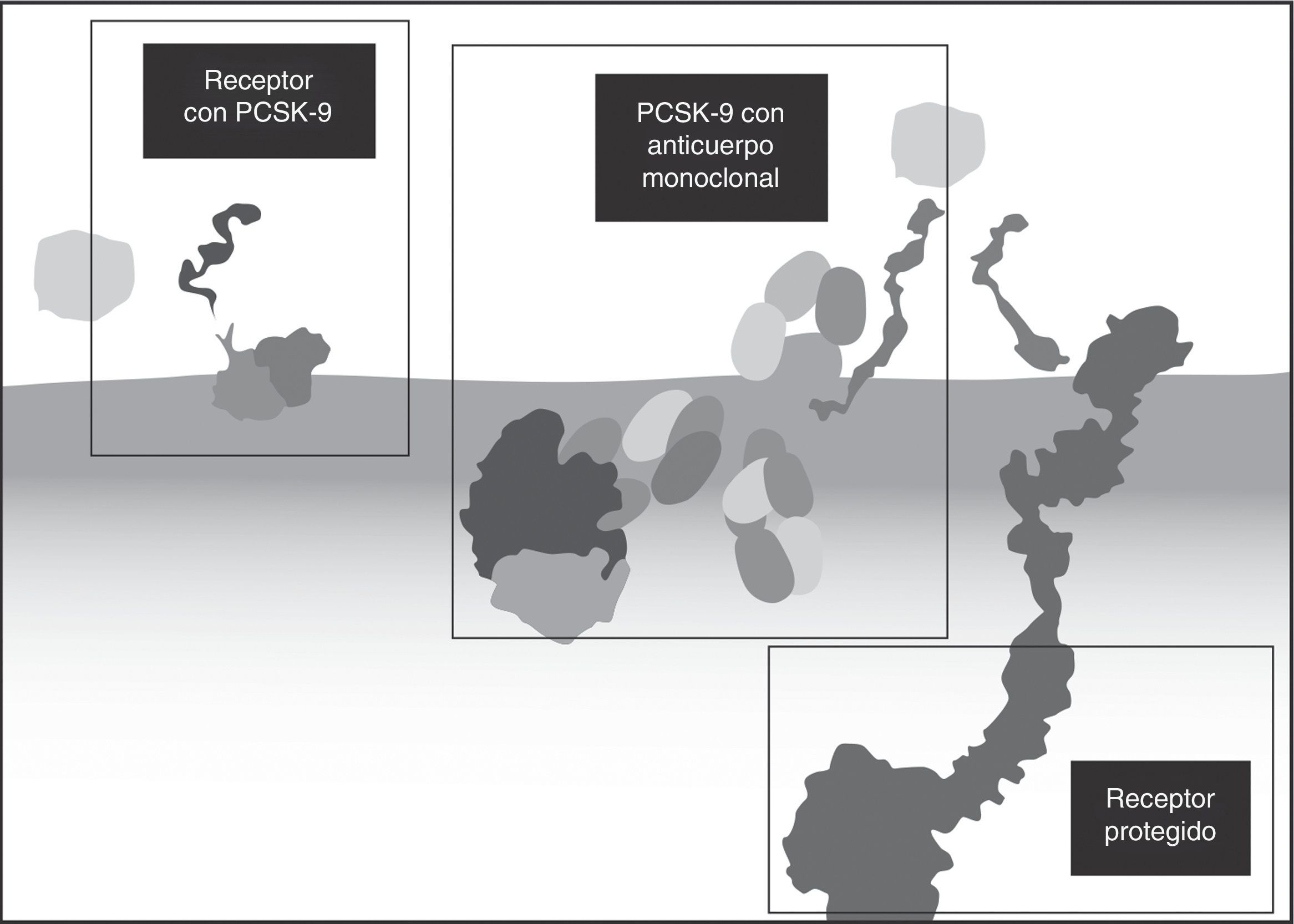
El efecto directo del anticuerpo monoclonal permite la inhibición de la función de la PCSK9, lo que aumenta el reciclaje de los receptores de LDL y secundariamente lleva a reducciones muy significativas de los niveles de cLDL en plasma34.
Desarrollo de las moléculas alirocumab y evolocumabEn las primeras fases, los anticuerpos se administraron de manera intravenosa para garantizar una biodisponibilidad completa y asegurar que las concentraciones plasmáticas fueran las ideales. Una vez evaluada y garantizada la función, se estudió la inyección subcutánea, que tiene como ventajas la mejor conveniencia para los pacientes, menores riesgos de reacciones en la infusión, menor costo y facilidades logísticas superiores.
Varias razones hacen que el perfil de seguridad de alirocumab y de evolocumab, los dos anticuerpos estudiados y aprobados, sea muy apropiado: se trata de anticuerpos completamente humanos, lo que disminuye la opción de generar reacciones inmunes (anticuerpos contra los anticuerpos) o de generar anticuerpos neutralizantes; el riesgo de interacciones medicamentosas es bajo, puesto que no tienen metabolismo hepático ni renal ni interactúan con el citocromo P45061,63. ni con otras proteínas de transporte. Adicionalmente, tienen alta especificidad por sus blancos terapéuticos y alta efectividad con dosis espaciadas (cada dos o cuatro semanas) y no penetran al sistema nervioso central (por el tamaño de la molécula, que es incapaz de atravesar la barrera hematoencefálica intacta)61. Como una ventaja adicional, importante en pacientes con enfermedad cardiovascular aterosclerótica, los anticuerpos monoclonales no bloquean los canales de potasio, de manera que no tienen efectos sobre el intervalo QT ni sobre la repolarización cardíaca35. La figura 5 muestra el mecanismo de acción, en el que el anticuerpo se une a la PCSK9 y evita que esta se adhiera al RLDL36.
Demostración de efectividad
Las dos moléculas cuentan con amplios programas de investigación, que les han permitido obtener resultados demostrados en diferentes situaciones: en terapia aislada, como adición a estatinas, en pacientes con hipercolesterolemia familiar homo o heterocigota, en reducción de placa, en eventos adversos neurocognoscitivos y finalmente en eventos duros (fig. 6).
Programa PROFICIO, con evolocumab
En particular, cabe destacar del programa PROFICIO con evolocumab y los estudios LAPLACE-237 y YUKAWA-2 en combinación con estatinas; en pacientes que no toleran las estatinas los estudios GAUSS-238 y GAUSS-3; como tratamiento único, MENDEL-39; en pacientes con hipercolesterolemia familiar heterocigota (HF He) RUTHERFORD-240 y TAUSSIG; y en pacientes con hipercolesterolemia familiar homocigota (HF Ho) TESLA y TAUSSIG.
En términos de estudios de efectividad y seguridad a largo plazo, los estudios de evolocumab produjeron resultados de gran interés:
- •
OSLER-241 (Open Label Study of Long TERm Evaluation Against LDL-C Trial-2): mostró reducción a un año de 61% en cLDL, comparado con terapia estándar, y disminución en la tasa de eventos cardiovasculares con un HR de 0,47 (IC95% 0,28 a 0,78).
- •
GLAGOV (GLobal Assessment of Plaque ReGression with a PCSK9 AntibOdy as Measured by IntraVascular Ultrasound)42: evaluó el efecto en placa aterosclerótica de pacientes llevados a cateterismo cardíaco y demostró la reducción de placa en quienes recibieron evolocumab.
- •
TAUSSIG43 (Trial Assessing Long Term USe of PCSK9 Inhibition in Subjects with Genetic LDL Disorders) en pacientes con HF homo y heterocigota: en un subanálisis de 106 pacientes con hipercolesterolemia homocigota, 34 recibían aféresis, se evaluó evolocumab, el cual logró reducción del 20,6% en el cLDL, que se mantuvo por 48 semanas, sin diferencias significativas con los pacientes en aféresis. Este estudio, junto con el TESLA, fue la base para la aprobación de la indicación de uso en HF Ho.
- •
DESCARTES44 (Durable Effect of PCSK9 Antibody CompARed wiTh PlacEbo Study) en pacientes con riesgo cardiovascular: mostró un perfil apropiado de seguridad, que no alteró las hormonas esteroideas ni gonadales ni produjo efectos adversos significativos, aún en niveles muy bajos de cLDL.
- •
FOURIER (Further Cardiovascular OUtcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk)45: sin duda, el más importante de los estudios hasta la fecha, puesto que demostró, en 27.564 pacientes tratados por 48 semanas, reducción en cLDL de 59% (de una media basal de 92mg/dl a 30mg/dl), así como en el desenlace primario (un compuesto de muerte cardiovascular, infarto de miocardio, ACV, hospitalización por angina inestable o revascularización coronaria) del 15% (HR 0,85, IC95% 0,79 a 0,92), y en el desenlace secundario (compuesto de muerte cardiovascular, infarto de miocardio o ACV) del 20% (HR 0,80, IC95% 0,73 a 0,88). Adicionalmente, no hubo diferencias significativas en términos de eventos adversos, incluyendo diabetes de aparición reciente y eventos neurocognoscitivos.
- •
EBBINGHAUS46: 1.204 pacientes del estudio FOURIER fueron seguidos por 19 meses y evaluados en busca de eventos adversos neurocognoscitivos; no se encontraron cambios en los desenlaces primarios (índices de funciones ejecutivas) ni en los secundarios (memoria de trabajo, memoria episódica o velocidad psicomotriz). Tampoco hubo diferencias cuando se analizaron los niveles de cLDL obtenidos.
Se hicieron estudios en HF He (ODYSSEY HF I y ODYSSEY HF II47, así como ODYSSEY HIGH HF48), en pacientes con niveles altos y muy altos de cLDL, con reducciones significativas en cLDL a pesar de recibir tratamiento de base con estatinas.
También en pacientes a quienes se trataba con la máxima dosis tolerada de estatinas, con o sin ezetimibe (ODYSSEY COMBO I49 y ODYSSEY COMBO II50), en los que se logró, con alirocumab, reducción significativa y sostenida de cLDL, sin efectos adversos importantes.
El estudio ODYSSEY LONG-TERM51 evaluó la terapia de alirocumab a largo plazo en pacientes con alto riesgo cardiovascular en quienes no se habían logrado las metas de cLDL a pesar de tratamiento hipolipemiante. Se obtuvieron reducciones significativas de cLDL sin eventos adversos. El estudio no estaba diseñado para evaluar desenlaces duros.
El ODYSSEY MONO52 evaluó alirocumab en monoterapia, comparado con ezetimibe en pacientes con hipercolesterolemia primaria y riesgo moderado. La reducción resultante, de 47,2%, fue claramente superior a la lograda con ezetimibe.
En el estudio ODYSSEY ALTERNATIVE53 se estudiaron alirocumab o ezetimibe en pacientes con intolerancia a las estatinas. Los resultados fueron superiores para alirocumab (reducción de cLDL de 45,0%).
Los estudios ODYSSEY OPTIONS I54 y OPTIONS II55 evaluaron comparaciones con ezetimibe agregado a tratamientos con atorvastatina o con rosuvastatina en diversas dosis. La mayoría (alrededor del 80,0%) de los pacientes, lograban las metas de cLDL cuando recibían alirocumab.
Por su parte, el estudio ODYSSEY OUTCOMES fue diseñado para estudiar desenlaces duros en aproximadamente 18.000 pacientes durante 5 años; está próximo a terminarse.
ConclusionesEvolocumab y alirocumab, los anticuerpos monoclonales específicos contra la PCSK9, ya aprobados y en uso clínico, han demostrado efectividad en la reducción del cLDL y en eventos clínicos; su efectividad se debe a la protección del receptor específico para LDL, que puede reciclar cientos de veces y cumplir su función de captación del colesterol. En los casos en los que haya producción de receptores, los anticuerpos monoclonales serán útiles como potentes reductores del cLDL.
Esta nueva adición al arsenal terapéutico, los anticuerpos monoclonales contra la PCSK9, es una herramienta terapéutica que puede ser de gran utilidad en los pacientes con niveles elevados de cLDL, en particular en casos de HF heterocigota, en algunos casos de HF homocigota y en personas con alto riesgo cardiovascular cuyos niveles no pueden ser controlados con dosis máximas toleradas de estatinas. Cabe aclarar que la dosis máxima tolerada de estatinas puede ser ninguna estatina, en los casos en los que haya verdadera intolerancia.
Esta nueva familia de medicamentos ya demostró efectividad (con evolocumab, estudio FOURIER) en la reducción de eventos duros (compuestos de muerte cardiovascular, infartos, ACV, angina o revascularización), y seguridad neurocognoscitiva.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.