


Los cambios epigenéticos inducidos por factores ambientales tienen cada día más relevancia en las enfermedades cardiovasculares. Uno de los componentes moleculares más observados en la hipertrofia cardiaca es la reactivación de los genes fetales causados por diversas patologías que incluyen obesidad, hipertensión arterial, estenosis valvular aórtica, causas congénitas, entre otras. A pesar de las múltiples investigaciones cuyo objetivo es obtener información acerca de los componentes moleculares de esta patología, su influencia en las estrategias terapéuticas es relativamente escasa.
En la actualidad se busca información acerca de las proteínas que modifican la expresión de los genes fetales que se reactivan en esta condición. La relación entre las histonas y el ADN tiene un control reconocido en la expresión de genes que son condicionados por el ambiente e inducen modificaciones epigenéticas. Las deacetilasas de histonas son un grupo de proteínas que han demostrado tener un papel importante en la diferenciación de la célula cardiaca y además pueden ser claros componentes en el desarrollo de la hipertrofia cardiaca. En este trabajo se revisan los conocimientos actuales sobre la influencia de estas proteínas y los posibles planes terapéuticos en la hipertrofia cardiaca.
Epigenetic alterations induced by environmental factors are more relevant each day for cardiovascular diseases. One of the most observed molecular components in hypertrophic cardiomyopathy is the reactivation of fetal genes caused by multiple conditions, including obesity, high blood pressure, aortic valve stenosis and congenital causes. Despite several investigations with the objective of obtaining information regarding molecular components of this condition, its influence in therapeutic strategies is relatively scarce.
Nowadays information is being searched about proteins that modify the expression of the fetal genes that reactivate with this condition. The relationship between histones and DNA has a recognised control in the expression of genes that are subject to the environment and induce epigenetic alterations. Histone deacetylases are a group of proteins that have revealed to play an important role in differentiation the cardiac cell and could be clear components in the development of hypertrophic cardiomyopathy. In this study current knowledge about the influence of these proteins and possible therapeutic plans for hypertrophic cardiomyopathy are revised.
De acuerdo con la última nota descriptiva del centro de prensa de la Organización Mundial de la Salud (publicada en enero de 2015), las enfermedades cardiovasculares son la principal causa de muerte en todo el mundo. Cada año mueren más personas por esta causa que por cualquier otra, incluido el cáncer. Se calcula que en 2012, a esta condición se le atribuyeron 17,5 millones de muertes, lo cual representa un 31% de todas las muertes registradas en el mundo1.
En la cardiopatía hipertrófica el corazón experimenta una respuesta adaptativa–por lo general reversible–a estímulos fisiológicos, como el ejercicio o el embarazo; esta respuesta se caracteriza por el aumento del tamaño de los cardiomiocitos, con el fin de bombear suficiente sangre para suplir los requerimientos hemodinámicos2.
Estudios anatómicos demuestran que el límite superior normal en cuanto al peso del corazón es alrededor de 450g para los hombres y 400g para las mujeres. En ambas condiciones, la relación entre el peso del ventrículo izquierdo y la estatura no debe exceder 36g/m2, por lo que el punto de corte establecido para esta condición por ecocardiografía3 es de 50g/m2. Por encima de este límite, es más probable que se trate de una hipertrofia mal-adaptativa o irreversible bajo condiciones que incluyen hipertensión crónica, infarto del miocardio, obesidad, diabetes y angina, entre otras4. En contraste con estas formas de hipertrofia secundarias al estrés miocárdico, existe otra entidad genética conocida como miocardiopatía hipertrófica, que también se asocia con un aumento del grosor de la pared ventricular izquierda más allá de la respuesta a cargas anómalas5,6; sin embargo, la fisiopatología de esta entidad es diferente y no se abordará en esta revisión.
Las tecnologías “ómicas” han facilitado el rápido progreso para comprender los determinantes genéticos y factores de riesgo implicados en la patogénesis de enfermedades de alta complejidad, incluidas las cardiovasculares7. Se sabe que estos fenómenos fisiopatológicos se encuentran mediados por perfiles fenotípicos y genotípicos en los que se destaca la capacidad notable de plasticidad del corazón adulto. Por lo tanto, la aplicación de enfoques biológicos contemporáneos permite dilucidar las vías celulares y moleculares que conducen a hipertrofia cardíaca, atrofia y alteraciones funcionales secundarias8,9.
Esta revisión se centrará en los avances recientes de los diferentes mecanismos epigenéticos centrados en la modificación de la cromatina, que pueden desencadenar una respuesta hipertrófica como adaptación a sobrecarga. El estudio de estos mecanismos moleculares representa un avance relevante para esclarecer los procesos subyacentes en las enfermedades cardiovasculares y por lo tanto blancos terapéuticos para prevención y control de las mismas.
Fisiopatología de la cardiopatía hipertróficaEl corazón de los mamíferos es una bomba inagotable capaz de suplir los requerimientos hemodinámicos y metabólicos de todo el organismo, llevando oxígeno y nutrientes a todos los tejidos10. En respuesta a la sobrecarga, el corazón se adapta entrando en un proceso de hipertrofia, con lo que se disminuye el estrés en las paredes ventriculares y logra mantener e incluso aumentar su función eyectora11.
En el adulto, el crecimiento cardiaco es mediado inicialmente por un aumento en el tamaño de los cardiomiocitos (hipertrofia) y no en el número de estos (hiperplasia). Aunque los cambios agudos en el gasto cardíaco son estimulados a través del sistema simpático, en presencia de alteraciones prolongadas del mismo se genera hipertrofia como respuesta. Esto puede ocurrir como resultado de la adaptación, similar al que se observa en hipertensión arterial crónica, infarto agudo del miocardio, causas genéticas (miocardiopatía), infección (miocarditis), enfermedad valvular cardíaca, trastornos tiroideos, obesidad, diabetes, envejecimiento, etc.11.
En cuanto al crecimiento no-patológico del corazón, se incluye la cardiogénesis fetal, el crecimiento postnatal cardiaco y el aumento modesto en el tamaño del corazón, lo cual puede ocurrir en ausencia de actividad contráctil. La configuración de las cuatro cámaras cardiacas se alcanza en el segundo trimestre de la gestación y continúa aumentando de tamaño para mantener el apoyo circulatorio necesario para el crecimiento del embrión9.
En roedores y en otros modelos experimentales, el crecimiento fetal depende en gran parte de la hiperplasia, hasta el nacimiento, donde la división celular disminuye de manera gradual12.
Cuando la hiperplasia desaparece, muchas células se someten a una ronda final de cariogénesis (división nuclear) sin citocinesis (división celular) produciendo así una mezcla de cardiomiocitos mononucleados y binucleados (el sincitio cardiaco). Entonces el crecimiento del ventrículo izquierdo supera al del ventrículo derecho durante el período postnatal temprano, fenómeno que se encuentra relacionado con la transición de la circulación fetal a la adulta en el corazón. En estas circunstancias el corazón tiene un aumento de hasta seis veces su masa inicial13,14. Las células cardiacas en el periodo posnatal en el humano no se dividen y la expresión de los genes propios de la división celular se detienen, es decir todos los genes fetales cardiacos bloquean su expresión.
En la cardiopatía hipertrófica se desarrollan procesos fisiológicos y patológicos9–11. Los patológicos obedecen a un aumento en la apoptosis celular, la remodelación cardiaca y la disminución en la función sistólica y diastólica, llevando así a una falla cardiaca, lo cual ocurre después de estímulos como la insuficiencia coronaria, el deterioro valvular e incluso la hipertensión arterial15,16.
En los humanos y en modelos animales, los cardiomiocitos ventriculares sometidos a hipertrofia patológica reactivan los genes que normalmente se expresan en altas concentraciones durante la vida fetal. Este “programa genético fetal” incluye genes como ANP y BNP, actina alfa esquelética e isoformas fetales de la cadena pesada de miosina. Adicionalmente, se retoma el comportamiento metabólico fetal disminuyendo la tasa de oxidación de ácidos grasos y aumentando la de oxidación de la glucosa17–19. Otros cambios fisiológicos celulares, incluyen la reorganización del sarcómero, las alteraciones en la homeostasis de calcio y las variables en la contractilidad y la relajación asociadas a muerte de los cardiomiocitos con subsecuente fibrosis y remodelación eléctrica20.
Los mecanismos moleculares que intervienen en el desarrollo de la cardiopatía hipertrófica están relacionados con procesos de mecano-transducción y vías de señalización mediados por los receptores acoplados a proteínas G, el receptor Janus Quinasa que son transductores de señal y activadores de la transcripción (JAK-STAT) y las proteínas quinasas activadas por mitógenos (MAPKs)9.
Si bien el efecto mecánico que genera la sobrecarga sobre el cardiomiocito es suficiente para generar hipertrofia, la mecano-transducción es un fenómeno crítico para el desarrollo de la cardiopatía hipertrófica patológica y fisiológica. El cardiomiocito detecta estos estímulos a través de complejos proteicos del sarcómero, integrinas del citoesqueleto, canales iónicos activados por el estiramiento y receptores activados por ligandos21,22.
Dichas señales activan la cascada de señalización que se traduce en un aumento en la síntesis de proteínas, acoplamiento miofibrilar, inducción de la transcripción de genes que aumentan el tamaño de los cardiomiocitos (hipertróficos) y angiogénesis. Adicionalmente, la activación de integrinas estimula al cardiomiocito23, para que libere angiotensina II y endotelina-1. Por otro lado, las catecolaminas se unen a receptores acoplados a proteínas G, activando la cascada de señalización a través de la fosfolipasa C produciendo liberación de calcio intracelular y activando el complejo calcio-calmodulina a través del factor nuclear de células T activadas (calcineurina-NFAT) y quinasas dependientes de calmodulina (CaMK)24.
Algunos factores de crecimiento, tales como el factor de crecimiento similar a la insulina tipo 1 (IGF-I), el de crecimiento de fibrobalastos (FGF) y el de crecimiento vascular endotelial (VEGF), se unen a receptores con actividad tirosina quinasa intrínseca en sus dominios citoplasmáticos. La fosforilación del receptor, que en el cardiomiocito, está acoplada a fosfoinositol 3 quinasa (PI3K), fosfolipasa C y GTPasas de la familia RAS, que actúan como traductores de señales, especialmente en el desarrollo de hipertrofia cardiaca por medio de la vía IGF-I-PI3-K, y adicionalmente, activa vías de señalización con el factor de crecimiento vascular 3 (VEGF 3) controlado por tres centros de señalización ERK1/2, el complejo mTOR, mientras que la AMP dependiente quinasa (AMPK), rige la reprogramación de la adaptación metabólica con el fin de aumentar la síntesis de proteínas25–27.
Así pues, la activación de ERK1/2 en el corazón conduce a hipertrofia cardiaca in vivo. Curiosamente esta activación produce una hipertrofia cardiaca concéntrica con función sistólica conservada sin evidencia de fibrosis, lo que sugiere que ERK1/2da como resultado una hipertrofia cardiaca compensada28,29.
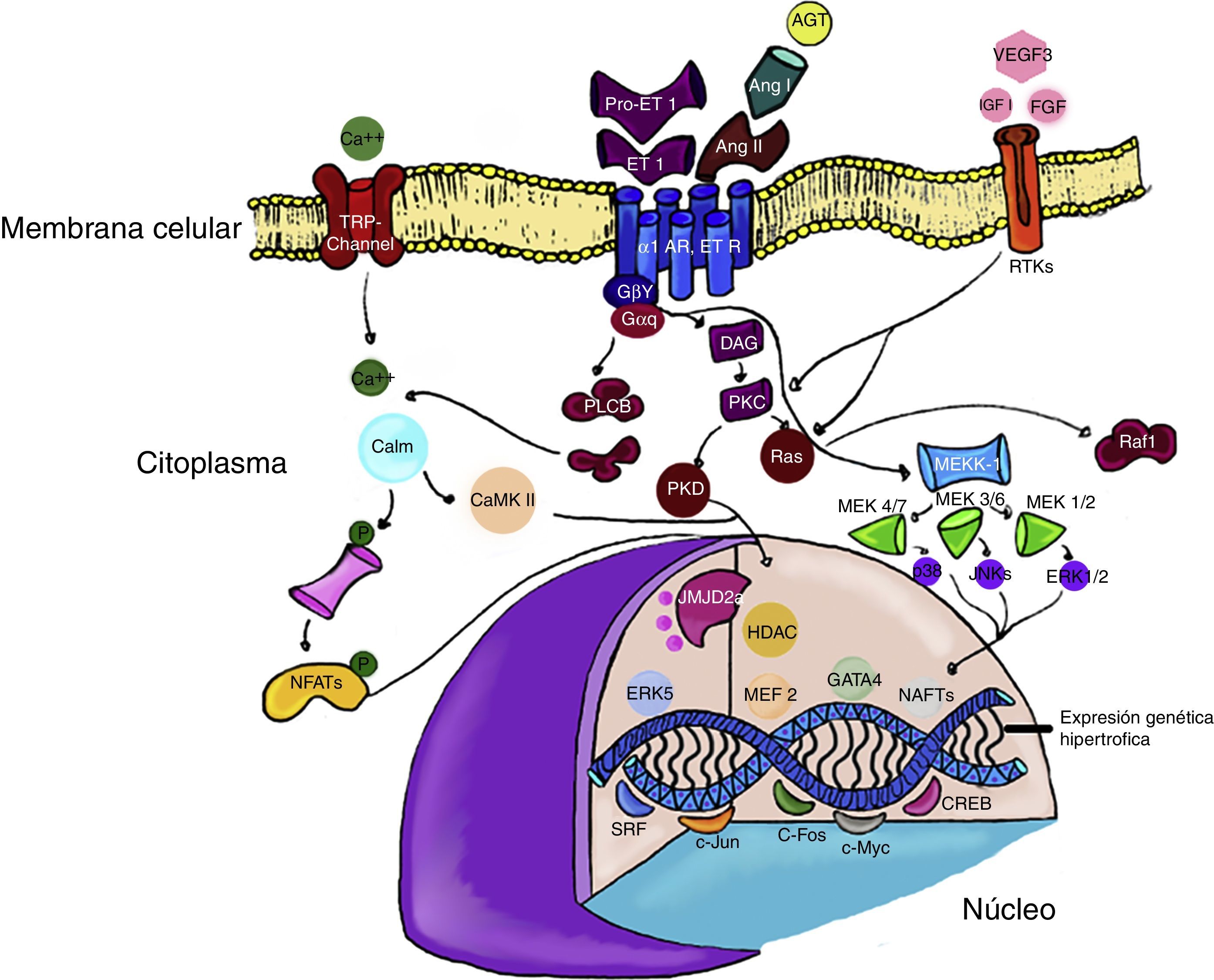
En contraste, la activación de ERK5 se asocia a hipertrofia concéntrica, excéntrica, miocardiopatía dilatada y muerte súbita. Estos fenotipos dispares demuestran como la activación de algunos modelos específicos de MAPK, puede conducir a diferentes formas de hipertrofia cardiaca y subraya la importancia de ampliar estudios en el tema30,31 (fig. 1).
 y angiotensina II (Ang II), pueden estimular la señalización mediante la diacilglicerol (DAG) por la vía de RAS y quinasas mitogénicas (MEKK). Por su parte, otros factores de crecimiento como factor similar a la insulina (IGF-1), y fibroblasto (FGF) fosforilan los receptores tirosina quinasa rTK) en la membrana celular. En la cromatina los factores de transcripción c-JUN y c-Fos pueden inducir la transcripción de genes específicos que inducen la hipertrofia cardiaca. Algunos modificadores de la transcripción, como las histonas deacetilasas (HDAC) o las demetilasas de histonas (JMJD2A), pueden alterar la expresión génica (explicación en el texto).")
Vías de señalización de la hipertrofia cardiaca. Los receptores para endotelina (ET-1) y angiotensina II (Ang II), pueden estimular la señalización mediante la diacilglicerol (DAG) por la vía de RAS y quinasas mitogénicas (MEKK). Por su parte, otros factores de crecimiento como factor similar a la insulina (IGF-1), y fibroblasto (FGF) fosforilan los receptores tirosina quinasa rTK) en la membrana celular.
En la cromatina los factores de transcripción c-JUN y c-Fos pueden inducir la transcripción de genes específicos que inducen la hipertrofia cardiaca.
Algunos modificadores de la transcripción, como las histonas deacetilasas (HDAC) o las demetilasas de histonas (JMJD2A), pueden alterar la expresión génica (explicación en el texto).
Este término se utilizó por primera vez para referirse a interacciones complejas entre el genoma y el medio ambiente involucradas en el desarrollo y la diferenciación del organismo. En la actualidad se conocen las diferentes modificaciones moleculares y los procesos heredables en el ADN, que son independientes de los cambios en la estructura del ADN y son más bien modificaciones enzimáticas. Estas últimas incluyen metilación del ADN, especialmente en la zona del promotor de los genes, modificaciones covalentes de las histonas, remodelación de la cromatina dependiente de ATP y regulación por RNA no codificante32,33.
Muchas de las modificaciones epigenéticas tienen la unidad básica de la cromatina, conocida como nucleosoma, que consiste de 146 pares de bases de ADN envueltos alrededor de un octámero de histonas y se compone de dos copias de cada una de las cuatro histonas del núcleo: H2A, H2B, H3 y H419. Los residuos de aminoácidos de las histonas, especialmente aquellos en las colas (extremo N-terminal), están sujetos a modificaciones postrascripción, muchas reversibles desde el punto de vista enzimático, como acetilación, metilación, fosforilación, ubiquitinación y SUMOilación3,20 proporcionando mecanismos de ajuste en respuesta a estímulos celulares34,35.
De esta manera, las histonas son reconocidas como elementos cruciales en la mediación de las adaptaciones fenotípicas del corazón durante los procesos fisiológicos o patológicos, así como en el desarrollo embrionario del mismo36,37. Por ejemplo, el mecanismo central de alteración de la estructura de la cromatina y el control de expresión de genes, es representado por la acetilación/deacetilación de histonas. La acetilación por histonas acetil-transferasas (HAT) hace que se pierda la interacción histona-ADN (relaja la cromatina y activa la transcripción) mientras que la deacetilación por histonas deacetilasas (HDAC) aumenta la interacción de histona-ADN (condensando la cromatina y reprimiendo la transcripción)38,39.
Las deacetilasas de histonas clase II (HDAC 4, HDAC 5, HDAC 7 y HDAC 9) son expresadas de manera significativa en el corazón, de ahí que responden a la señalización molecular limitando el crecimiento de los cardiomiocitos y la hipertrofia.
Las HDAC II reprimen la transcripción de genes mediante la interacción con el factor potenciador de miocitos 2 (MEF2), así que se cree que parte de su función represiva se relaciona con mantener la región del promotor en un estado deacetilado40,41.
Por su parte, la metilación de las histonas tiene un papel relevante en la regulación de la expresión génica y forma parte del sistema de memoria epigenética que regula el destino celular y la especificidad funcional42,43.
Metilación vs. demetilación de histonasUn aspecto importante en la regulación de la epigenética es la diafonía entre las diferentes modificaciones postransduccionales como fosforilación, ubiquitinación, SUMOilación, acetilación y metilación44. Originalmente se creía que la metilación de la lisina, en contraste con otras modificaciones, era estática y que solo podría ser modificada al ser reemplazada por una nueva histona45. De hecho la di[2]-metilación y tri[3]-metilación de las histonas son características de la heterocromatina, donde la actividad de expresión de genes se encuentra anulada. Sin embargo, estos residuos diferencialmente metilados, sirven como sitio de acoplamiento para proteínas efectoras y modificadoras de la cromatina (afecta el plegamiento de orden superior), lo que conduce a diversas respuestas fisiológicas tales como la represión o activación transcripcional y la reparación del ADN dependiendo del residuo de lisina metilado46.
De esta forma la metilación de las “colas” de las histonas, es un proceso dinámico regulado por dos clases de enzimas: las histonas metiltransferasas (HMT) y las histonas demetilasas (HDMT). Estas enzimas pueden generar activación o represión de la transcripción dependiendo del residuo de aminoácido metilado, especialmente lisina o arginina, y del grado de metilación (mono, di, o trimetilación)47–49.
Adicional al descubrimiento de la metiltransferasa de las histonas, la demetilasa específica de lisina 1 (LSD1) y la familia de proteínas del dominio JMJC han cambiado el panorama de la epigenética48.
En forma reciente se ha observado que la demetilasa de histonas JMJD2A/KDM4A tiene un papel importante en la fisiología del corazón. El aumento de la función de JMJD2A incrementa la hipertrofia cardiaca en respuesta a la presión por la exposición a sobrecarga, mediado por la demetilación de los residuos de la histona 3-lisina 9 (H3K9 y la activación de genes como el péptido natriurético auricular (ANP) y ventricular o de tipo B (BNP), efecto probablemente realizado en conjunto con HDAC450.
Epigenética: las demetilasas y su papel en el desarrollo fisiológico y patológico del corazónPara el desarrollo del corazón de los vertebrados se requiere la diferenciación simultánea de varios tipos celulares. Existen cinco grandes etapas del desarrollo del corazón identificadas en ratones, incluyendo la etapa de crecimiento cardiaco (E7.75), la formación lineal de tubo cardíaco (E8.0), la iniciación y la formación de las cámaras cardiacas (E9.5), la maduración y separación de las mismas (E12.5) para finalmente dar formación a las válvulas a partir de E12 hasta el nacimiento3.
Dentro de este proceso se conocen dos poblaciones de células progenitoras cardíacas, las cuales se pueden encontrar en la etapa creciente cardiaca y se identifican como el primer campo cardiaco (FHF) y una región más medial, las cuales se encuentran adyacentes entre sí, denominada segundo campo cardíaco (SHF)3.
El primer campo cardiaco da lugar al tubo cardiaco inicial que está destinado a convertirse en el miocardio del ventrículo izquierdo. Las células progenitoras del segundo campo cardiaco permanecen indiferenciadas hasta que se unen con el tubo cardiaco para formar el ventrículo derecho y el tracto de salida. Este proceso es controlado por las células de la cresta neural para formar posteriormente cojines endocárdicos en el tracto de salida. Este desarrollo está mediado por un grupo de factores trascripcionales cardiacos (Mesp1, Isl1, Nkx2.5, Mef2c, Tbx1, Gata4, Foxa2/c1/c2/h1 and Hand2) y factores de crecimiento (Fgf8/10, Wnt3a/5a/11, Bmp2/4/7, Shh)51. El factor de transcripción Mesp1 representa el marcador más precoz de los progenitores cardiovasculares incluyendo los derivados del primer y segundo campo cardiaco. De tal manera que tanto Mesp1 como Mesp2 son necesarios para establecer el mesodermo embrionario que da origen al corazón52.
Mutaciones en la expresión normal de estos factores durante el desarrollo del corazón han demostrado que constituyen la presencia de cardiopatías congénitas tales como defectos septales, malformaciones atrioventriculares o ausencia del desarrollo del corazón derecho o izquierdo53.
Las histonas poseen proteínas que le confieren su carga positiva, por lo que atraen ADN el cual posee una carga negativa; por tanto, una vez acetiladas, el grupo acetil neutraliza las cargas positivas y fuerza a la histona para que pierda el agarre con el ADN haciéndolo más disponible22.
En sentido molecular, se caracteriza por una alteración dramática de la expresión genética asociada con el aparato contráctil y el metabolismo. Genes fetales como βMHC que normalmente se expresan en el corazón embrionario y fetal, son reactivados en los cardiomiocitos adultos mientras que las isoformas adultas como αMHC son reprimidas13.
Nuevos estudios indican que el balance en la metilación de la lisina es fundamental en el mantenimiento de la integridad del genoma, la regulación de genes y la evasión del cáncer, demostrándose así una relación entre los defectos en la regulación genética de la KMT y KDMS asociados con el desarrollo embrionario, el envejecimiento, el cáncer y los trastornos neurológicos54.
Así, esta amplia gama de modificaciones epigenéticas de la cromatina ofrece un punto de integración para la multitud de señales que inciden sobre los cardiomiocitos, permitiendo la estabilización, modulación y expresión de genes de acuerdo con estímulos específicos3.
Potencial diagnóstico y terapéuticoLa evidencia disponible en la actualidad demuestra que las alteraciones en los procesos de metilación y demetilación de las histonas, pueden conducir a enfermedades cardiovasculares de alto impacto en adultos, como lo es la hipertrofia cardiaca y de esta manera favorecer complicaciones como la falla cardiaca.
Algunos estudios recientes manifiestan el papel de diferentes demetilasas de las histonas en el proceso de diferenciación de la célula cardiaca. Es así como JMJD3a y UTX, que tienen la capacidad de demetilar la histona 3 en los residuos de lisina 27 (H3K27), tienen un papel relevante en la diferenciación de la célula cardiaca55. Adicionalmente, JMJD2A/KDM4A ha demostrado un rol preponderante en el desarrollo de las células musculares, y promueve la activación transcripcional del gen MyoD mediante el cual contribuye a la diferenciación del músculo esquelético56,57.
En un estudio previo, Zhang et al. desarrollaron células cardiacas con mayor expresión y ausencia de la expresión del gen de JMJD2a/KDM4A, observando que las células que tenían inhibición completa de JMJD2A presentaron menor respuesta hipertrófica ante el efecto de sobrecarga provocado por la constricción aórtica transversal. Por otro lado, aquellos con sobreexpresión de este presentaron una respuesta hipertrófica cardiaca exagerada34. Este grupo observó que el mecanismo más probable mediante el que JMJD2A regula la actividad de los genes fetales e induce hipertrofia cardiaca es la regulación del promotor de FHL1, un importante componente para la mecano-transducción en el corazón, induciendo hipertrofia cardiaca a través del factor de respuesta sérico (SRF) y el miocardio para generar un aumento en la expresión de ANP58. Por lo tanto, estos estudios respaldan el potencial como diana terapéutico del KDM4A en pacientes con falla cardiaca.
ConclusiónEn esta revisión se han expuesto las implicaciones que tienen modificaciones epigenéticas en el desarrollo de la hipertrofia cardiaca. Se espera que estudios más profundos logren caracterizar el mecanismo de los procesos mediante los cuales la variación de las histonas afecta la expresión de genes fetales y puedan desarrollarse estrategias terapéuticas para la hipertrofia y la falla cardiaca.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.