






Las enfermedades priónicas son un grupo de enfermedades neurodegenerativas raras y rápidamente progresivas que causan síntomas neuropsiquiátricos diversos. Estas enfermedades se describieron hace más de 200 años, y con el tiempo se reconoció que los animales eran portadores de esta alteración; sin embargo, hasta finales de los noventa este problema conmocionó Europa, ya que para entonces la enfermedad había cruzado la barrera de especie y podía afectar al hombre. La polémica fue mayor al atribuirse la alteración a una proteína con capacidad infecciosa. El proceso patológico común se caracteriza por la conversión de la proteína priónica celular normal (PsPc) a una forma anómala y patológica (PrPSc). En el ser humano se han clasificado como padecimientos idiopáticos, hereditarios o adquiridos por la exposición a material exógeno con contenido priónico. La manifestación neurológica más sobresaliente de las prionopatías consiste en la aparición de una demencia rápidamente progresiva asociada a mioclonías y ataxia cerebelosa, además de síntomas extrapiramidales. Los síntomas psiquiátricos ocurren en etapas tempranas de la enfermedad y su presencia, además de la valoración de factores de riesgo, puede contribuir al diagnóstico oportuno de este síndrome. Clásicamente los síntomas psiquiátricos se han agrupado en tres categorías: síntomas afectivos, alteraciones de la motricidad y síntomas psicóticos. Este tipo de manifestaciones suele aparecer durante un periodo prodrómico previo a los signos neurológicos y consiste en la aparición de aislamiento social, ideas delirantes, irritabilidad/agresión, alucinaciones predominantemente visuales, ansiedad y depresión, así como otros menos frecuentes. El diagnóstico definitivo requiere de un estudio post mortem. La posibilidad de que un número mayor de casos pueda ocurrir en los próximos años o que en muchos pacientes no se haya considerado el diagnóstico es una realidad. En opinión de los autores de este trabajo, los psiquiatras debemos tener conocimiento de los síntomas de esta enfermedad. El objetivo de esta investigación es evaluar las alteraciones neuropsiquiátricas presentes en las prionopatías y, en particular, determinar si las manifestaciones psiquiátricas en conjunto integran un cuadro clínico que apunte al diagnóstico de estas enfermedades, aunque en primer término se revisan aspectos taxonómicos, patogénicos y patológicos. Como elemento agregado en este trabajo, los autores hacen algunas consideraciones diagnósticas basadas en la evidencia científica disponible hasta el momento. Los descriptores controlados aplicados a la búsqueda bibliográfica son los utilizados para indexación de artículos científicos en las bases de datos. Las bases de datos electrónicas utilizadas fueron PubMed y EMBASE, aunque también se empleó PsycInfo. Los descriptores empleados son: enfermedades priónicas, trastornos psicóticos, depresión, trastornos afectivos, patología, clasificación, proteína priónica, historia, manifestaciones neurológicas y manifestaciones psiquiátricas. Los criterios de selección de material fueron cualitativos. Como conclusión y con base en la extensa bibliografía revisada, los autores plantean que el periodo en que hay más evidencia de alteraciones en la esfera mental se denomine «fase de síntomas psiquiátricos», la cual puede extenderse por algunos meses (hasta 4). Los síntomas afectivos son los más característicos de dicha fase. Como conclusiones, se considera que la identificación de estos síntomas en un paciente con factores de riesgo de sufrir esta enfermedad contribuiría a la identificación temprana del padecimiento y normaría qué pautas seguir ante la sospecha del diagnóstico de este grupo de trastornos, sobre todo con la idea de mejorar la calidad de vida de estos pacientes.
Prion diseases are a group of rare and rapidly progressive neurodegenerative conditions that may cause neuropsychiatric symptoms. This group of diseases has been described since the 18th century, but they were recognized decades later, when it became clear that the humans were affected by infected animals. There was controversy when the problem was attributed to a single protein with infective capacity. The common pathological process is characterized by the conversion of the normal cellular prion protein into an abnormal form. In humans, the illness has been classified as idiopathic, inherited and acquired through exposure to exogenous material containing abnormal prions. The most prominent neurological manifestation of prion diseases is the emergence of a rapidly progressive dementia, mioclonus associated with cerebellar ataxia and also extra pyramidal symptoms. Psychiatric symptoms occur in early stages of the illness and can contribute to timely diagnosis of this syndrome. Psychiatric symptoms have traditionally been grouped in three categories: affective symptoms, impaired motor function and psychotic symptoms. Such events usually occur during the prodromal period prior to the neurological manifestations and consists in the presence of social isolation, onset of delusions, irritability/aggression, visual hallucinations, anxiety and depression, and less frequent first-rank symptoms among others. Definite diagnosis requires post mortem examination. The possibility that a large number of cases may occur in the next years or that many cases have not been considered with this diagnosis is a fact.
In our opinion, psychiatrists should be aware of symptoms of this disease. The main objective of this research consisted of assessing the correlation between this disturbance and neuro-psychiatric symptoms and particularly if this psychiatric manifestations integrate a clinical picture suggestive for the diagnosis of these diseases, but firstly reviewed taxonomic, pathogenic and pathological aspects. The authors of this project also added an element in relation to some diagnostic considerations based on scientific evidence. For the search controlled descriptors applied to the research for indexing scientific articles in databases were used. The electronic data bases used were PubMed, EMBASE and also PsycInfo. The descriptors were prion diseases, psychotic disorders, depression, mood disorders, pathology, classification, prion protein, history, neurological manifestations, and psychiatric manifestations. The selection criteria for the material were qualitative. To conclude, and based on the extensive literature review, the authors propose that the period where the evidence is more robust for mental impaired is named “psychiatric symptoms phase, which can be extended for a few months, being the psychiatric affective symptoms the most characteristic of this phase. In conclusion, we considered that the identification of these symptoms in a patient with risk factors for developing the disease will contribute to the early identification, and would regulate the guidelines in suspected diagnosis of this group of disorders. The intention is provide a better quality of life to the sick people.
Falta título en inglés y un abstract legible (¿no hay una versión que no tenga todas las palabras pegadas?)
cita 1: ¿Será Prions. Proc Natl Acad Sci U S A. 1998;95:13363-83? No encuentro la publicación del ‘82 con ese título.
IntroducciónLas enfermedades priónicas (también conocidas como prionopatías o encefalopatías espongiformes transmisibles) son un grupo de afecciones neurodegenerativas descritas por primera vez en el ganado ovino hace más de 200 años. A finales de la década de los años noventa esta enfermedad conmocionó al mundo tras sus primeras apariciones en seres humanos en el continente europeo1. El diagnóstico definitivo de este padecimiento se basa en el examen neuropatológico post mortem del cerebro, pues no existe una prueba clínica diagnóstica sensible y específica en vida.
El diagnóstico probable se alcanza en función de los criterios clínicos establecidos por la Organización Mundial de la Salud (OMS), los cuales se basan en la combinación de características clínicas compatibles y un electroencefalograma característico o por la detección de una proteína neuronal que se acumula en el líquido cefalorraquídeo (LCR) en situaciones de destrucción neuronal importante, la cual se le conoce como proteína 14-3-3. Los estudios de imagen servirán de apoyo, pero solo son importantes para descartar otras enfermedades. Según lo encontrado en diferentes reportes, durante la fase inicial de la enfermedad predominan síntomas neuropsiquiátricos que pueden contribuir a la identificación temprana del padecimiento.
Considerando el interés en el tema, así como su trascendencia y su relevancia, y tomando en cuenta la importancia de que el psiquiatra en su actividad clínica cotidiana esté familiarizado con enfermedades potencialmente generadoras de manifestaciones psiquiátricas, aun cuando estos padecimientos no sean muy frecuentes, se estimó pertinente explorar el estado actual de los conocimientos sobre el particular. En respuesta a este interés, se efectuó una revisión de la literatura. El objetivo general de este trabajo es conocer diversos aspectos relacionados con esta enfermedad teniendo como centro principal las manifestaciones neuropsiquiátricas.
Para cumplir con este objetivo, se tomó la decisión de dividir este trabajo en tres apartados. El primero, muy breve, aborda tópicos muy generales de tipo histórico. La segunda sección incluye aspectos de definición y clasificación, y acto seguido se examina lo concerniente a las características de la proteína priónica celular, así como la patogenia y la patología. Tras esto, se establece una aproximación a los tipos y las características de las manifestaciones neuropsiquiátricas, no sin antes señalar brevemente elementos clínicos de tipo neurológico. Finalmente, en el tercer apartado se analizan detalles relacionados con la fenomenología de las expresiones conductuales y psiquiátricas de las prionopatías. Con el propósito de que no sea solamente una revisión narrativa y descriptiva, los autores plantean algunas opiniones en el apartado de discusión con base en la bibliografía sobre algunas consideraciones diagnósticas tomando en cuenta diversos elementos clínico-psiquiátricos y, básicamente, según el tiempo de evolución o curso clínico de la enfermedad.
MétodosDe acuerdo con la práctica basada en la evidencia, se efectuó una búsqueda computarizada de información utilizando la estrategia PICO (paciente o problema, intervención, comparación o control y outcomes o resultados). La idea de esta investigación surgió para responder el interrogante de si las prionopatías son generadoras de alteraciones neuropsiquiátricas. Según la estrategia PICO, se describieron todos los componentes relacionados con el problema identificado y se estructuró la pregunta de investigación: ¿Cuáles son las características más sobresalientes de las prionopatías desde ángulos tan diversos pero tan complementarios como los taxonómicos, clínicos y fenomenológicos (principalmente neuropsiquiátricos), así como patogénicos y patológicos?
Acto seguido se inicio la búsqueda bibliográfica seleccionando en primer lugar como términos de búsqueda descriptores relacionados con cada una de las etapas de la estrategia PICO. Los descriptores controlados aplicados son los utilizados para indexación de artículos científicos en las bases de datos. Se utilizó el MeSH (MEDLINE/PubMed) y el EMTREE (EMBASE). Las bases de datos electrónicas utilizadas son precisamente PubMed y EMBASE, aunque también se empleó PsycInfo. Asimismo se manejaron los operadores boleanos (delimitadores) representados por los términos AND, NOT y OR, con los que se realizaron combinaciones de palabras. Los descriptores empleados son: enfermedades priónicas, trastornos psicóticos, depresión, trastornos afectivos, patología, clasificación, proteína priónica, historia, manifestaciones neurológicas y manifestaciones psiquiátricas. Los criterios de carácter cualitativo para seleccionar el material fueron: artículos originales, revisiones, metanálisis o editoriales en idioma inglés o español, publicados de 1980 a abril del 2014; estudios cuantitativos, y trabajos con metodología científica y editoriales con aportaciones concretas. Se excluyeron trabajos que repetían información o con evidentes sesgos metodológicos. Del material recabado (110 artículos), se seleccionaron finalmente 66 referencias, en función de su relevancia y su aportación teórica o práctica. Artículos anteriores al periodo establecido se consideraron en consenso entre los autores solamente si eran contribuciones que habían trascendido a pesar del tiempo transcurrido, siempre y cuando se apegaran a los criterios establecidos.
Antecedentes históricosLos registros más antiguos de las enfermedades por priones corresponden a las observaciones efectuadas a mediados del siglo xviii1 sobre una enfermedad que afectaba al ganado ovino en muchos países de Europa y que por sus síntomas se denominó scrapie en Inglaterra, traberkrankheit des schafes en Alemania y tremblante du mouton en Francia2.
A principios de 1920, Creutzfeldt y Jakob3 describieron los primeros casos de encefalopatía espongiforme en el ser humano, por lo que dicha enfermedad recibió la denominación de enfermedad de Creutzfeldt-Jakob (CJD, por sus siglas en inglés).
En 1936, los trabajos de Cuillé et al4 demostraron el carácter contagioso de la enfermedad y su largo periodo de incubación, lo que llevaría a Sigurdsson5 en 1953 a denominarlas «infecciones lentas». Trabajos posteriores demostraron la transmisión del scrapie a otras especies como el visón, las cabras y algunos roedores6–8. Seis años después, Hadlow9 relacionó el scrapie con una enfermedad endémica en los nativos de Nueva Guinea, llamada enfermedad de Kuru. Más tarde, Gibbs et al10 transmitieron experimentalmente la CJD a los chimpancés, y en los años siguientes reprodujeron dicho procedimiento en otras especies animales.
Estas estrategias y las observaciones derivadas de ello permitieron clasificar las encefalopatías como espongiformes transmisibles; ejemplos de estas son el síndrome de Gerstmann-Sträussler-Scheinker en humanos11 y la encefalopatía transmisible del visón o las encefalopatías de los cérvidos silvestres (chronic wasting disease [CWD])12.
En 1960, el grupo de Gajdusek10 terminó por demostrar su transmisibilidad y en 1982 Prusiner13 descubrió al agente patógeno, los priones, y demostró que se trataba de partículas puramente proteicas sin ácido nucleico. Por estos hallazgos le fue otorgado al autor el Premio Nobel de Medicina en 1997.
Durante la última década se han descubierto nuevas formas de encefalopatías espongiformes transmisibles del gato14, el antílope15 y el ciervo eland16; de estas, la encefalopatía espongiforme bovina es la de mayor publicidad porque se ha demostrado que se transmite a los seres humanos.
DefiniciónLas prionopatías constituyen un conjunto de enfermedades neurodegenerativas producidas por acumulación de una isoforma anormal de la proteína priónica celular (PrPc). La PrPc es fundamental para la transmisión sináptica en el sistema nervioso central17. Esta evidencia ha dado lugar a que diversos autores consideren estas enfermedades alteraciones en la conformación proteica normal de la membrana plasmática celular17,18.
Este grupo de enfermedades también recibe el nombre de encefalopatías espongiformes transmisibles (ETT) y afectan tanto a humanos como a animales19. En el ser humano, según lo observado por Arranz et al20, se pueden clasificar como padecimientos esporádicos/idiopáticos, hereditarios o adquiridos por exposición a material exógeno que contenga priones21. En la tabla 1 se describe con más detalle esta clasificación.
Clasificación de las prionopatías20,21
| Esporádica/idiopática/clásica | Enfermedad de Creutzfeld-Jakob esporádica (sCJD) |
|---|---|
| Adquiridas (exógenas) | Enfermedad de Creutzfeld-Jakob iatrogénica (iCJD) |
| Variante de la enfermedad de Creutzfeld-Jakob (vCJD) | |
| Kuru | |
| Genética/hereditaria | Enfermedad de Creutzfeld-Jakob familiar (fCJD) |
| Insomnio familiar letal | |
| Síndrome de Gerstmann-Sträussler-Scheinker (GSS) |
Una de las manifestaciones más comunes y devastadoras asociadas a las prionopatías22 es la aparición de una demencia rápidamente progresiva evidente hasta en un 62% de los casos.
Características de la proteína priónica celularLa PrPc es una glucoproteína de la membrana plasmática compuesta por 253 aminoácidos y un puente disulfuro, con un peso molecular de 33-35 kDa; se codifica en el ser humano normalmente por un gen situado en el brazo corto del cromosoma 20 humano (20p12). Entre los diferentes polimorfismos de este gen, destaca el situado en el codón 129, que puede codificar para valina (V) o metionina (M), lo cual resulta relevante para el desarrollo de la enfermedad. Investigaciones recientes han vinculado la mutación de otros genes como E200K, D178N, P102L y V210I en la patogenia de formas esporádicas de este grupo de enfermedades23. Estudios realizados en Alemania han asociado la mutación del codón 178 de la PRNP con el insomnio familiar letal (IFL), una forma hereditaria de enfermedad priónica con un fenotipo variado y caracterizada por mioclonías, desorientación espacial y alucinaciones, así como disfunción neurovegetativa que, al igual que en el resto de estas enfermedades culmina con una demencia rápidamente progresiva24. Esta proteína se encuentra presente en el ser humano desde el inicio de la embriogénesis, y en la edad adulta se encuentra distribuida en diferentes tejidos como músculo esquelético, cardiaco y linforreticular, pero es principalmente abundante en el sistema nervioso central, sobre todo en las neuronas del cerebro y la médula espinal, así como en las células gliales. Al gen que codifica para PrPc se lo denomina «gen de la proteína priónica» (PRNP), y su polimorfismo marca el tipo de enfermedad por priones que padece el paciente25. El desarrollo de las prionopatías depende de los cambios que esta proteína sufra. La proteína priónica y la forma patológica (PrPSc) comparten la misma estructura primaria, esto es, la misma secuencia de aminoácidos; la diferencia fundamental radica en su conformación.
La proteína se pliega en hélices alfa principalmente, y posee escasas láminas beta; este cambio conformacional es lo que confiere a la proteína priónica de capacidad autoduplicarse, además de resistencia a diversos agentes fisicoquímicos26,27. El patrón de glucosilación y el fragmento resistente a proteasas definen las distintas cepas de PrPSc, de las cuales hasta el momento se han caracterizado cinco tipos27.
Patogenia y patologíaLas enfermedades priónicas del ser humano son biológicamente únicas y, como ya se ha comentado, pueden dividirse en heredadas, esporádicas y adquiridas20,21,28. Aproximadamente el 85% de ellas ocurren de manera esporádica29 y alrededor del 15% de estas enfermedades se han asociado con mutaciones autosómico dominantes en el gen PRNP30.
Las formas adquiridas se han confinado a situaciones extremadamente raras e inusuales, como en la transmisión de priones a través de hormonas tiroideas derivadas de cadáveres humanos, implantes de duramadre o corneales y con el uso concomitante de electrodos electroencefalográficos; esta constituiría la transmisión por vía intracerebral31.
Según diversas investigaciones32, la vía cerebral resulta ser la más efectiva para la transmisión de priones, lo cual es lógico teniendo en cuenta que el tejido nervioso es el órgano diana principal de estas enfermedades. Esta vía de acceso evade los sistemas inmunológicos innatos y adquiridos, así como la barrera hematoencefálica.
En la CJD iatrogénica (iCJD), la transmisión ocurre vía neuroquirúrgica o tras los injertos de duramadre31. La transmisión oral de priones ha causado grandes epidemias; el caso más común sería el del Kuru, una enfermedad priónica transmitida por los rituales de canibalismo practicados por las tribus del norte de Papúa-Nueva Guinea33,34.
Otro ejemplo más reciente es la variante de CJD causada por el consumo de carne contaminada de bovinos con encefalopatía espongiforme en Reino Unido y algunos países de América35,36. Estudios recientes37,38 han demostrado que los priones se transmiten con eficacia entre algunas especies de animales como ratones y cérvidos (mamíferos rumiantes) a través de partículas en aerosol o gotas de Flügge.
Hallazgos como los de Budkay et al39 muestran que no se aprecian anormalidades macroscópicas en cerebros de pacientes con enfermedad priónica, pero en el examen microscópico se puede encontrar cambios histopatológicos característicos consistentes en vacuolización y degeneración neuronal, lo que da a la materia gris una apariencia microvacuolar o «espongiforme», además de la proliferación de células de la astroglía. Otro hallazgo que resulta incluso más frecuente en estas enfermedades es la astrogliosis y microgliosis40.
En revisiones como la de Wadsworth41 se describe como característica especial la falta de respuesta inflamatoria en linfocitos. El diagnóstico de certeza de una infección por priones se hace demostrando inmunorreactividad a la PrPSc o más específicamente con la detección bioquímica de esta en la sustancia gris por técnicas de inmunotransferencia; todo esto post mortem, como ya se ha mencionado39.
Otras formas de enfermedades priónicas se caracterizan por el depósito de placas de amiloide compuestas de agregantes insolubles de PrPSc24,40. Estas placas son notablemente características en el Kuru y el síndrome de Gerstmann-Sträussler-Scheinker. Los hallazgos de carácter histopatológico en CJD son constantes y se distinguen de otras enfermedades priónicas; sobresalen las placas amiloideas con gran número de PrPSc y un tejido adyacente a la placa de apariencia microvacuolar que da a las placas una morfología que semeja una flor42.
Manifestaciones neurológicasSegún lo señalado previamente, lo más sobresaliente es la presencia de una demencia rápidamente progresiva que se asocia a mioclonías y ataxia cerebelosa en un 90% de los pacientes43. Otros síntomas extrapiramidales incluyen temblor, coreoatetosis y parkinsonismo (60%). En el 50% de los pacientes hay signos de afección piramidal43,44.
La evolución es invariablemente fatal. La CJD es la forma más común de encefalopatía espongiforme. La media de edad al inicio es alrededor de los 65 años, con una media de supervivencia de 4 meses45. Los pacientes con enfermedad secundaria a injertos son típicamente más jóvenes y pueden desarrollar manifestaciones con predominio de la ataxia sobre la demencia. En la tabla 2 se señalan los criterios determinados por la Organización Mundial de la Salud (OMS) para el diagnóstico de la CJD esporádica46,47.
Criterios de la OMS diagnósticos de sCJD46,47
| Certeza diagnóstica | Características |
|---|---|
| Confirmado | Estudio post mortem: neuropatología o inmunohistoquímica |
| Probable | Demencia rápidamente progresiva y al menos dos de las siguientes características: mioclonías, signos de afección visual o cerebelosa, signos piramidales o extrapiramidales, mutismo acinético |
| Posible | Electroencefalograma anormal, proteína 14-3-3 positiva en líquido cefalorraquídeo, duración de la enfermedad < 2 años |
Datos obtenidos de la Unidad Nacional de Vigilancia de la CJD en Edimburgo (Reino Unido) reportan una serie de diferencias entre la forma esporádica y una nueva variante de dicho padecimiento (vCJD) (tabla 3)48,49.
Características clínicas que describen las diferencias entre la enfermedad de Creutzfeldt-Jakob esporádica y la variante48,49
| Variables | Creutzfeldt-Jakob esporádica | Variante de la enfermedad de Creutzfeldt-Jakob |
|---|---|---|
| Edad de inicio (años) | 65 (15-94) | 26 (12-74) |
| Duración (meses) | 4 (1-74) | 13 (6-39) |
| Presentación típica | Demencia progresiva, ataxia y mioclonías | Alteraciones psiquiátricas y del comportamiento |
Los valores expresan media (intervalo).
El fenotipo clínico de la vCJD es relativamente distinto de su forma esporádica. Las principales características corresponden a una mayor duración de la enfermedad, edad más joven al momento de la muerte (16-40 años) y el predominio de síntomas psiquiátricos en las etapas tempranas de la enfermedad50. Sin embargo, la poca especificidad de estos síntomas dificulta el reconocimiento temprano.
Según los hallazgos de Will et al51 en una población de 33 casos de vCJD confirmados, el primer especialista involucrado en la valoración inicial de los pacientes en más de la mitad de los casos de su muestra fue un psiquiatra. Asimismo los autores señalaron que el tiempo transcurrido desde el inicio de la enfermedad hasta la primera consulta fue de alrededor de 8 meses.
En esos estudios, los principales síntomas psiquiátricos fueron aislamiento, ideas delirantes persecutorias, irritabilidad/agresión, alucinaciones predominantemente visuales, síntomas afectivos como ansiedad y depresión y otros menos frecuentes, como síntomas del primer rango de Schneider (pensamiento sonoro, oír voces que discuten o dialogan entre sí, oír voces que comentan las propias acciones, robo e inserción del pensamiento), ideación suicida y olvidos frecuentes.
Al considerar dichos síntomas –como indican algunos autores52,53 en sus respectivos trabajos–, el problema clínico pareciera ser compatible con el diagnóstico de diversos trastornos afectivos, como depresión mayor, trastorno afectivo bipolar u otras alteraciones del comportamiento e incluso esquizofrenia.
Por otra parte, los escasos especificidad, sensibilidad y valores predictivos de los estudios de gabinete, como resonancia magnética o electroencefalograma, así como los análisis sanguíneos o de LCR, resultan ser poco útiles para el diagnóstico en etapas tempranas de la enfermedad. Brown et al54 señalan que los síntomas de déficit neurológico, característico de la demencia por priones, podrían no aparecer hasta pasadas algunas semanas o incluso meses, lo que complica la identificación inicial y la evolución de la enfermedad.
Otros expertos55 llegan a considerar como patognomónica la asociación de síntomas neurológicos y psiquiátricos y hallazgos característicos en el electroencefalograma. Señalan el enlentecimiento generalizado con ondas agudas irregulares de 0,5-1/s, y con una duración de 200-600ms, pero este patrón solo aparece a la mitad o en la fase final de la enfermedad.
En el análisis realizado por Wall et al56 en una muestra de 126 pacientes con diagnóstico de CJD, el 92% de los participantes presentaron al menos una manifestación psiquiátrica durante el curso de la enfermedad, y el 86% la presentó en los primeros 100 días de iniciado el padecimiento (etapa temprana). En esta etapa, el 79% de los pacientes presentaron alteraciones del ciclo sueño-vigilia, particularmente insomnio. Los síntomas psicóticos aparecieron en el 42% de los casos, en los que se incluyó una difusa gama de fenómenos alucinatorios y delirantes que iban desde delirios de persecución y paranoia hasta alucinaciones visuales, aunque también auditivas.
Otros autores reportan una mayor incidencia de delirios celotípicos51,53. En más de la mitad de los casos (77%), los síntomas psicóticos se presentaron en la etapa temprana. Los síntomas depresivos, como se ha señalado ya, no solo forman parte de la fase temprana, sino que también se manifiestan en la fase prodrómica del trastorno. Entre las manifestaciones afectivas, se incluyen afecto deprimido, aislamiento, hiporexia y pérdida de peso, lo cual no se atribuye a los trastornos motores de las fases tardías.
Menos frecuente es la presencia de fatiga, anhedonia y apatía, y se reportan ansiedad y «nerviosismo» hasta en un 21% de los pacientes del estudio56.
Otra manifestación psiquiátrica comúnmente reportada es agitación psicomotriz y conducta violenta, pero este hallazgo es similar a lo reportado en otros tipos de demencia57; no obstante, sería importante ampliar esta información, ya que habitualmente estas dos circunstancias son motivo de atención psiquiátrica en los servicios de urgencias de hospitales psiquiátricos y generales. Los diferentes estudios revisados51–56 señalan la aparición de al menos un síntoma psiquiátrico durante el curso de esta enfermedad; de estos, al menos el 25% de los síntomas ocurren en la fase prodrómica. Este padecimiento se puede confundir con una amplia variedad de trastornos psiquiátricos, particularmente afectivos, debido a los cambios en el comportamiento observados al inicio de la enfermedad.
La falta de respuesta al tratamiento y una mejoría únicamente sintomática podrían ser elementos de sospecha de la enfermedad para orientar el diagnóstico. La identificación de alteraciones cognitivas asociadas a los síntomas psiquiátricos incrementa la posibilidad de un trastorno orgánico subyacente. Ante la presencia de un cuadro clínico compatible con encefalopatía espongiforme, la primera consideración sin lugar a dudas debe ser la sCJD.
Fenomenología de las alteraciones conductuales y psiquiátricas de las enfermedades priónicas
Se han observado síntomas psiquiátricos y alteraciones conductuales desde los primeros casos de enfermedades priónicas publicados. Desde 1990, los síntomas de la esfera psiquiátrica forman parte de los criterios diagnósticos establecidos por la OMS para identificar la enfermedad58. Por desgracia, pese a los grandes esfuerzos realizados para identificar y entender esta enfermedad, no se ha logrado obtener información suficiente sobre prevalencia, historia natural y fenomenología de estas manifestaciones.
En los últimos años, los síntomas psiquiátricos y las alteraciones del comportamiento asociados a los trastornos demenciales han adquirido mayor relevancia clínica y científica. Hay una serie de retos que obstaculizan el estudio de los síntomas conductuales y psiquiátricos en las enfermedades priónicas, en comparación con las demencias más comunes. La poca frecuencia con que aparecen y el curso rápidamente progresivo de la enfermedad en combinación con la tendencia diagnosticar en fases tardías del padecimiento son factores que dificultan los estudios prospectivos.
Con base en los hallazgos de estudios previos, parecería conveniente clasificar los síntomas psiquiátricos y las alteraciones conductuales propias de este grupo de enfermedades en tres categorías principales: síntomas psicóticos, alteraciones en la motricidad y alteraciones del estado de ánimo51–54,56,59.
Los síntomas psicóticos pueden asociarse con agitación psicomotriz o alteraciones del comportamiento o no. Un síntoma común en este grupo de enfermedades es las alucinaciones visuales, que a su vez pueden correlacionarse con otras alteraciones como la agnosia visual60,61. Se ha descrito la presencia de ilusiones en un amplio número de pacientes. Algunos estudios sobre la fenomenología de este suceso lo describen como una fragmentación o distorsión desagradable de objetos y caras, «como si se tratara de un Picasso»59.
En cuanto al contenido de las alucinaciones, la mayoría reporta zoopsias y figuras humanas. En algunos casos se han descrito alucinaciones multimodales como ver, oír, sentir e incluso oler animales59. En estos pacientes y en estadios avanzados de la enfermedad, se observó una franca actitud alucinada con perplejidad y expresión de miedo en el rostro. Resulta importante mencionar que una cantidad importante de pacientes parece cursar con alucinaciones durante etapas tempranas de la enfermedad. Otro síntoma que indica psicosis en las prionopatías es las ideas delirantes, que suelen ser simples y relacionadas con persecuciones, robo o infidelidad58.
En un estudio de Cooper et al62, con una muestra de 204 pacientes con sCJD, 6 sujetos presentaron al inicio de la enfermedad únicamente alteraciones visuales, caracterizadas por pérdida de la agudeza visual y distorsiones, mientras que otros 29 presentaron alucinaciones e ilusiones asociadas a otros síntomas de la enfermedad durante la etapa temprana. La agitación y las alteraciones conductuales parecen ocurrir frecuentemente aun en ausencia de psicosis. La descripción típica, según diversos autores59,62, incluye irritabilidad, conducta desafiante y hostilidad.
En algunos pacientes, y sobre todo durante fases tempranas, las alteraciones conductuales consisten en signos de frontalización que incluyen desinhibición e impulsividad62. En estadios avanzados de la enfermedad, la agitación psicomotriz parece ser más reactiva ante estímulos sensitivos, y esta coexiste con mioclonías. Otros autores51–54 asocian las alteraciones de la motricidad a una respuesta exagerada a cualquier estímulo sensorial; esto es particularmente común en fases avanzadas de la enfermedad.
Algunos pacientes han manifestado que en determinados momentos cursan con alteraciones del humor, particularmente síntomas depresivos «clásicos», que son relativamente frecuentes al inicio63. Otras manifestaciones habituales son la tendencia al aislamiento y el retraimiento64. Los pacientes en fases intermedias de la enfermedad tienen mayor tendencia a cursar con labilidad emocional, apatía y abulia, al tiempo que comienzan a sufrir deterioro de la expresión del lenguaje y la función ejecutiva frontal65,66. Los síntomas afectivos en etapas finales de la enfermedad son poco característicos.
Discusión y conclusionesHistóricamente, las enfermedades priónicas han sido descritas como enfermedades inusuales, pero existe la posibilidad de que un número mayor de casos pueda ocurrir en los próximos años o que en muchos casos no se haya considerado el diagnóstico. Por ello, en opinión de los autores de este trabajo, los psiquiatras debemos tener conocimiento de los síntomas de esta enfermedad, tenerla en mente como parte del diagnóstico diferencial ante afecciones compatibles con cuadros demenciales de rápida progresión y para los que la mayoría de las veces no se logra identificar factores desencadenantes o causales.
También se estima necesario ser clínicamente escrupulosos, ante la dificultad que plantea el diagnóstico de estos padecimientos en particular durante las etapas tempranas, sin olvidar por supuesto que él dictamen definitivo es post mortem. La mayoría de las características clínicas de las enfermedades priónicas resultan de la pérdida irreversible de las funciones mentales y no remiten una vez que se inician. Las alteraciones conductuales y psiquiátricas son comunes en todos los tipos de enfermedad por priones aunque, como ya se ha mencionado, hay una heterogeneidad significativa entre los diferentes tipos de la enfermedad58.
Estos síntomas en general representan una carga significativa de morbilidad para los pacientes y sus cuidadores, pero también se debe considerar que fácilmente pueden pasar inadvertidos, a menos que los psiquiatras tratantes consideren esta eventualidad. Como se establece en la propedéutica médica, el primer paso para identificar un síntoma es indagar acerca de él.
En la mayoría de los casos reportados en la literatura actual, gran cantidad de los pacientes (25%) manifiestan al menos un síntoma psiquiátrico durante el curso de la enfermedad, que en su mayoría aparecen durante la fase prodrómica.
Ciertos factores de riesgo pueden orientar la búsqueda de síntomas específicos de este tipo de trastorno, entre otros los de índole sociodemográfica y cultural, como el consumo de carne contaminada en regiones endémicas o los rituales de canibalismo, así como factores identificados más recientemente, como el antecedente de una intervención quirúrgica del sistema nervioso central, sobre todo cirugías con aplicación de aloinjertos, y el no menos importante papel de la predisposición genética.
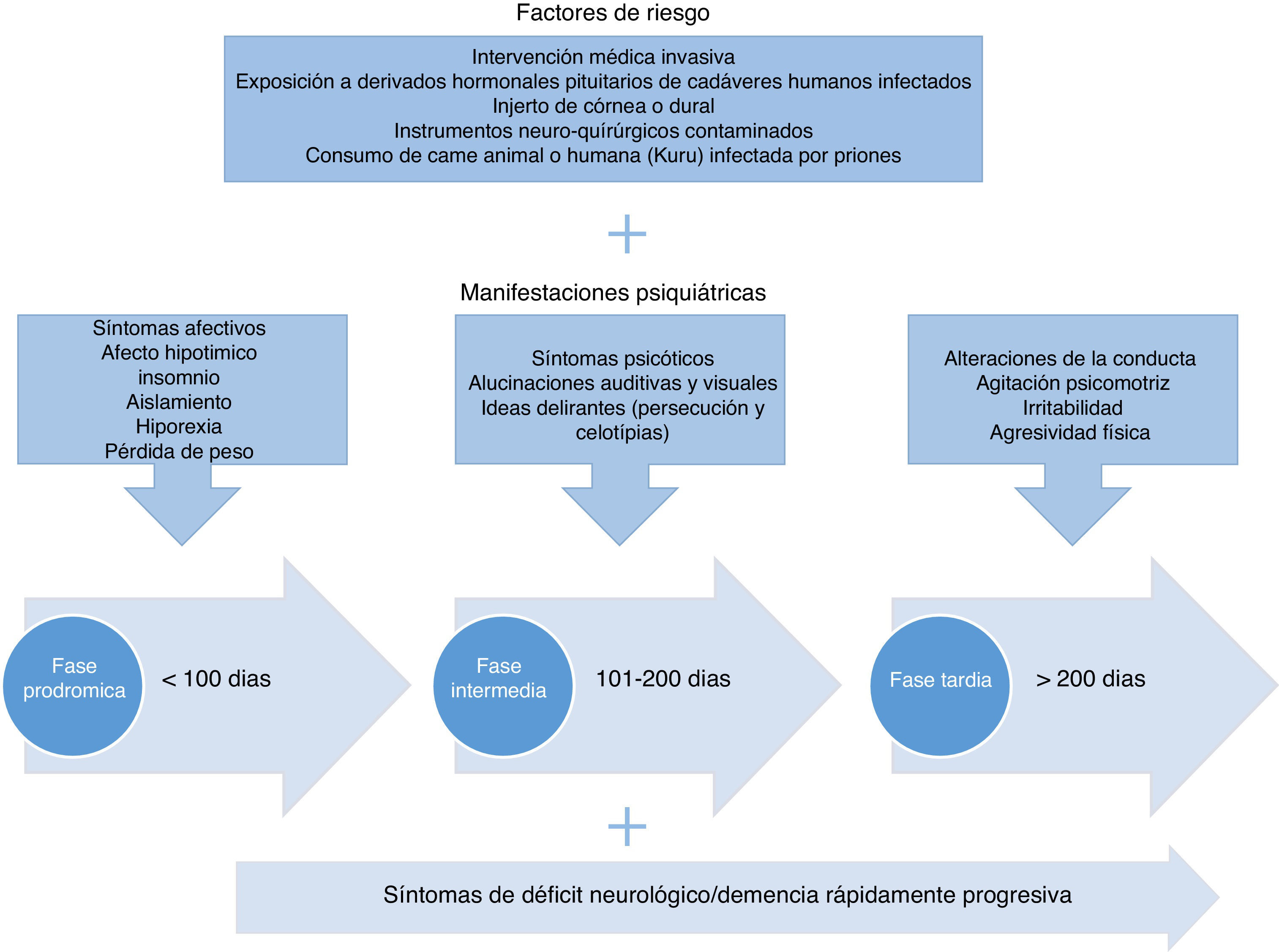
Para fines de la propuesta de este trabajo, y tomando en cuenta los hallazgos descritos, se plantea que el periodo con más evidencia de alteraciones en la esfera mental se denomine «fase de síntomas psiquiátricos», que puede extenderse algunos meses. En la figura 120,21,28,30–34,59,61,65 se describen los síntomas compatibles con enfermedad por priones, categorizándolos en síntomas afectivos y psicóticos y alteraciones de la motricidad acordes con el tiempo de instauración.

La aparición y la identificación de un deterioro cognitivo de rápida evolución agregado a los síntomas psiquiátricos puede incrementar la posibilidad de una alteración cerebral subyacente, lo que aunado a los factores de riesgo ya comentados puede orientar a que se considere la enfermedad priónica entre los diagnósticos diferenciales. Si bien es cierto que establecer un diagnóstico es un proceso imperfecto, la comprensión y el conocimiento de las diversas enfermedades pueden disminuir el grado de incertidumbre en la situación clínica.
En el caso de enfermedades que no se resuelven espontáneamente o no responden a tratamiento, sospecharla y analizar los síntomas a través del seguimiento pueden constituirse en métodos de referencia, de ahí que tener en mente las prionopatías no sea un ejercicio árido, vano o inútil. Consideramos que el caso del ébola es un claro ejemplo de que actualmente hay que valorar todas las posibilidades. Aunque no existe tratamiento específico para las enfermedades priónicas, la calidad de vida del paciente y sus cuidadores se podría beneficiar con el oportuno tratamiento sintomático de las manifestaciones neuropsiquiátricas comentadas.
Es factible mejorar estos síntomas, particularmente en casos graves de agitación psicomotriz y/o cuadros de psicosis, mediante el manejo farmacológico u otro tipo de intervención. Algunos datos indican que la mejoría de los síntomas psiquiátricos puede ser sustancial sin intervención específica, pero esto deberá quedar a consideración del psiquiatra tratante. Es un requisito agregado tomar en cuenta el riesgo-beneficio de la prescripción de fármacos.
La monitorización constante de los casos identificados y la intervención temprana en pacientes con sospecha de esta enfermedad pueden minimizar los factores que empeoran su curso clínico, que ya es letal en sí mismo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
