




We report the case of a 70-year-old male with chronic abdominal pain, who presented with increased intensity of the pain, and was initially diagnosed and treated for acute biliary pancreatitis. However, the symptoms persisted after hospital discharge, and he was subsequently studied with cholangio-MRI, abdominal CT scan, and ERCP, which revealed dilation of the biliary tract and a mass in the head of the pancreas. An excisional biopsy of the lesion showed chronic inflammation with severe sclerosing fibrosis and a lymphoplasmacytic infiltrate. A diagnosis of autoimmune pancreatitis was made, and the patient was initiated on azathioprine with good clinical response.
Presentamos el caso de un varón de 70 años con dolor abdominal crónico, que se presenta con aumento de este; se le diagnostica y se lo trata inicialmente como cuadro de pancreatitis biliar. A pesar del manejo inicial, los síntomas persisten después de la primera hospitalización, y se estudia con tomografía computarizada de abdomen, colangiorresonancia y colangiopancreatografía retrograda endoscópica, estudios en los cuales se detecta dilatación de la vía biliar y masa en la cabeza del páncreas. Se llevó a cabo biopsia excisional, que demuestra inflamación crónica con intensa fibrosis e infiltrado linfocitario. Se hace diagnóstico de pancreatitis autoimmunitaria y se inicia azatioprina, con adecuada respuesta.
Autoimmune pancreatitis is a rare disease that presents with clinical, serologic, and histologic characteristics of a defined autoimmune process that usually responds to the use of steroids.1 Yoshida introduced the term in 1995, replacing others that did not properly define the disease such as primary chronic pancreatitis, sclerosing chronic pancreatitis, duct destructive chronic pancreatitis, and sclerosing lymphoplasmacytic pancreatitis.2–6
Most information comes from Japan, where the number of documented cases has increased, probably due to improvements in diagnostic techniques and greater awareness of the disease. Cases have been reported in Europe, USA, and South Korea, which suggest that autoimmune pancreatitis has a worldwide prevalence. This disease may present as an isolated primary entity or it can be associated to other autoimmune diseases such as primary sclerosing cholangitis, primary biliary cirrhosis, retroperitoneal fibrosis, rheumatoid arthritis, sarcoidosis and Sjögren's syndrome.7,8
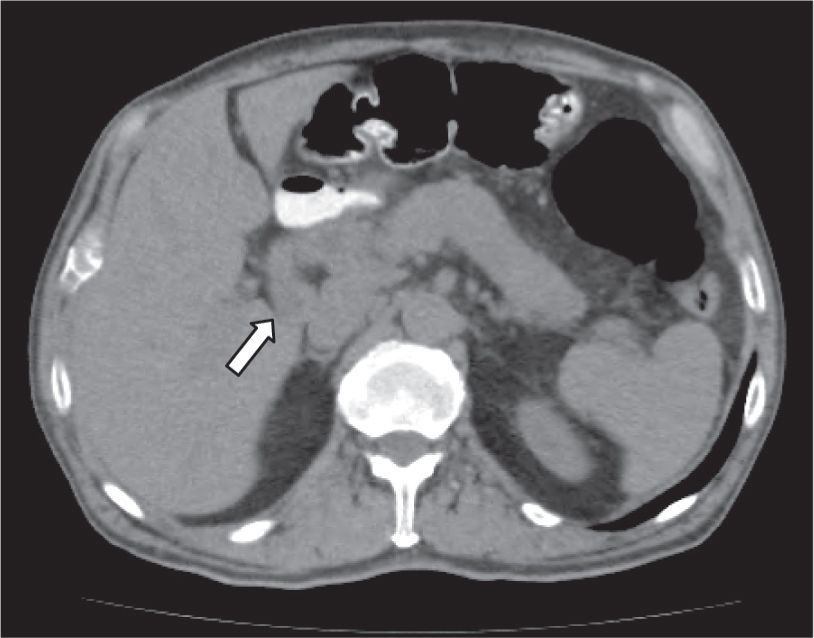
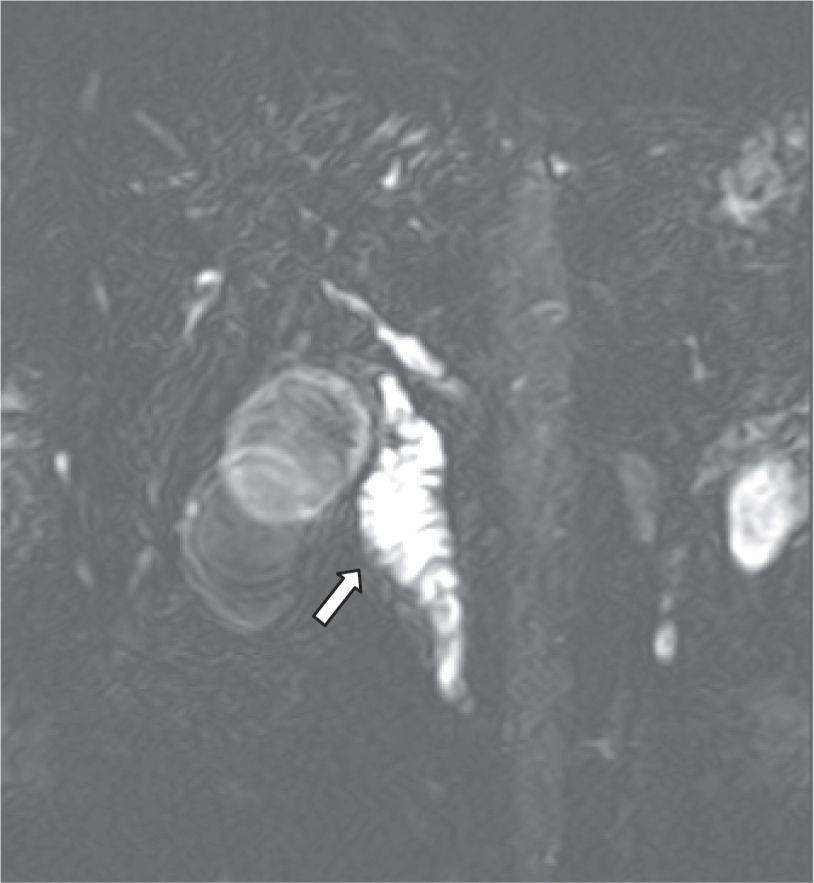
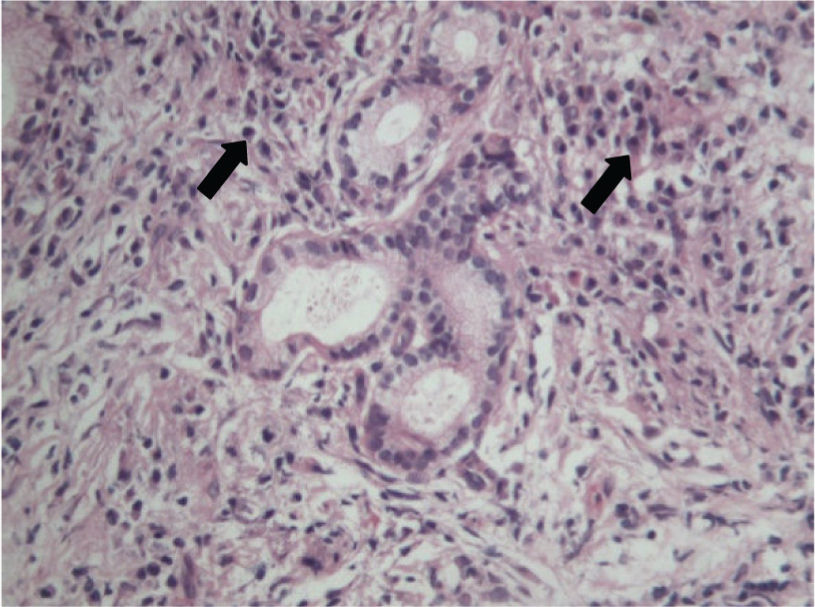
Case reportWe report the case of a 70-year-old male with a 10 month history of upper abdominal pain, jaundice, dark urine, pale stools, night sweats, generalized pruritus and weight loss of 28kg and no history of autoimmune disease. An initial diagnosis of biliary pancreatitis was made, and an endoscopic retrograde cholangiopancreatography (ERCP) was attempted, without success. Labs showed normal transaminase levels, direct hyperbilirubinemia, and elevated levels of alkaline phosphatase (972UI/L), amylase (174U/L) and lipase (1314U/L). Hepatobiliary ultrasound showed a distended gallbladder with thickened walls, dilated intra and extra-hepatic ducts, a 17mm dilation of the common bile duct, and a distal image that suggested biliary sludge associated with a diffuse growth of the pancreas. A new ERCP was attempted without success so a computed tomography (CT) scan was performed. The scan showed dilation of the biliary tree and a mass in the head of the pancreas (figure 1). The main differential diagnoses that were initially considered were cholangiocarcinoma and pancreatitis (Balthazar B). Subsequently, a cholangio-magnetic resonance imaging (MRI) revealed an obstruction of the biliary duct at the level of the intra-pancreatic tract, which could correspond to a multi-focal cholangiocarcinoma (figure 2). The patient was finally taken to surgery for an exploratory laparotomy, and frozen section biopsy revealed a chronic inflammatory process. Further histological studies showed destruction of glandular architecture with severe sclerosing fibrosis and lymphoplasmacytic infiltrates (figure 3). The patient was started on azathioprine, with good response.
")
")

Since autoimmune pancreatitis is a rare disease, its exact frequency is unknown. The prevalence has been estimated between 3.8 and 11% in patients with chronic pancreatitis, with a male-female ratio of 2.85.1,9,10. Most patients are diagnosed with the disease at age 55 or older, and up to 50% of them present with concomitant diabetes mellitus.9 In the United States, approximately 2.5% of the pancreaticoduodenectomies that were performed for masses that were diagnosed as cancer, and 23% of those that were done for suspected benign lesions, resulted in a histological diagnosis of autoimmune pancreatitis.11,12 This disease has been associated with other autoimmune diseases such as Sjögren's syndrome (12.2%), inflammatory bowel disease (7.6%), and retroperitoneal fibrosis (5.6%).13
Pathophysiological mechanisms described for autoimmune pancreatitis include both cellular and humoral immunity. Commonly, histological studies reveal lymphoplasmacytic infiltrates within the pancreatic lobules and ducts with a predominance of CD4+ and CD8+ lymphocytes.14 Patients with autoimmune pancreatitis may test positive for the following antibodies: anti-nuclear antibodies (ANAs), rheumatoid factor, lactoferrin, carbonic anhydrase II, and anti-smooth muscle antibody.14,15 These patients can also present with hypergammaglobulinemia and elevation of IgG, particularly IgG4, where even a systemic syndrome consisting of elevation of IgG4 and inflammation of the pancreas, salivary glands, biliary ducts and thyroid has been described.16–23 Finally, there also appears to be an association with DRB1*0405-DQB1*0401 HLA types.24
Clinical presentation may vary but there are two main patterns (table 1). Type 1 autoimmune pancreatitis presents in adults older than 60 years of age with obstructive jaundice and enlarged pancreas. The histological findings include lymphoplasmacytic infiltrate with elevation of IgG4. Type 2 autoimmune pancreatitis also presents with obstructive jaundice, but is associated to extensive destruction of the pancreatic ducts, usually presents in middle-aged patients, there is no elevation of IgG4, and is greatly associated with inflammatory bowel disease.25
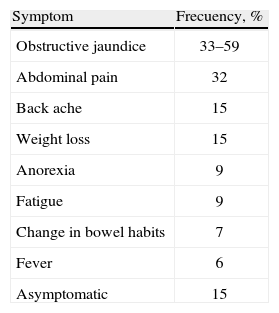
Autoimmune pancreatitis presents with chronic abdominal pain accompanied by acute, chronic or recurrent pancreatitis.14 Up to 85% of patients present with a pancreatic mass that might be mistaken for carcinoma or lymphoma with stenosis pancreatic ducts.15–17 Up to 80% of patients may develop exocrine dysfunction of the pancreas, and almost 77% may have altered endocrine functions.26 The diagnosis of autoimmune pancreatitis should always be considered in patients that present with symptoms that suggest cholangitis or inflammation of the pancreas, especially when there is history of autoimmune disease or recurrent episodes of pancreatitis. Even though it is a rare disease, its opportune diagnosis and treatment limit the progression of the disease, and can reduce the need of surgery and further complications.19,20
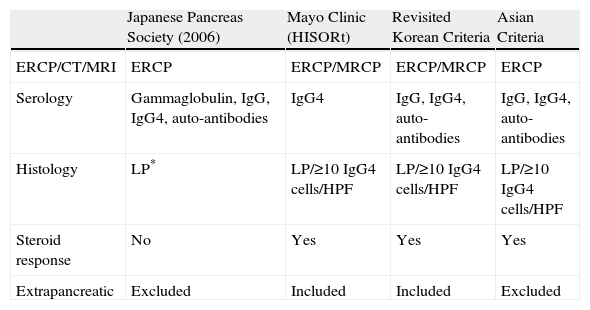
Different groups have proposed various diagnostic criteria for the disease (table 2).22,27–29 The most accepted are those established by the Japanese Pancreas Society (table 3).30
Comparison of diagnostic criteria for autoimmune pancreatitis
| Japanese Pancreas Society (2006) | Mayo Clinic (HISORt) | Revisited Korean Criteria | Asian Criteria | |
| ERCP/CT/MRI | ERCP | ERCP/MRCP | ERCP/MRCP | ERCP |
| Serology | Gammaglobulin, IgG, IgG4, auto-antibodies | IgG4 | IgG, IgG4, auto-antibodies | IgG, IgG4, auto-antibodies |
| Histology | LP* | LP/≥10 IgG4 cells/HPF | LP/≥10 IgG4 cells/HPF | LP/≥10 IgG4 cells/HPF |
| Steroid response | No | Yes | Yes | Yes |
| Extrapancreatic | Excluded | Included | Included | Excluded |
LP: lymphoplasmacytic sclerosing pancreatitis.
2006 Japanese Pancreas Society Criteria
| 1. Radiological evidence (US, CT-scan or MRI) of diffuse or segmental narrowing of the main pancreatic duct with irregular walls and diffuse or localized enlargement of the pancreas |
| 2. Elevation of serum gammaglobulin, IgG or IgG4, or positive auto-antibodies such as rheumatoid factor or antinuclear antibodies |
| 3. Marked interlobular fibrosis with lymphocytic and plasmacytic periductal infiltrate and, occasionally, lymphoid follicles within the pancreas |
CT: computed tomography; MRI: magnetic resonance imaging; US: ultrasounds.
Diagnosis: presence of criterion number 1 plus criterion number 2, 3 or both.
The main histological finding is lymphoplasmacytic sclerosing pancreatitis.32,33 The inflammation may compromise the peripancreatic fat, the pancreatic parenchyma and ducts, the veins (obstructive phlebitis), and the common bile duct.
Abdominal ultrasound normally reveals a “sausageshaped” diffuse enlargement of the pancreas with low echogenicity, and the common bile duct may appear dilated with thickened walls.34 CT scan usually shows an enlarged gland with parenchymal fibrosis and a capsule-like rim surrounding the lesions. This finding is very specific for autoimmune pancreatitis.23,27 MRI findings include diffuse enlargement of the pancreas with low signal in T1-weighted images and also a capsule-like rim in delayed-enhancement T2-weighted images.23,31 ERCP usually shows narrowing of the common bile duct and sometimes the intrahepatic ducts as well.19,22,35
Usually there is hypergammaglobulinemia (43%), elevation of serum IgG (62–80%), IgG4 (68–92%), ANAs (40–64%) and RF (25%).36 Other markers that may be found are: antibodies against carbonic anhydrase II (55%), lactoferrin (75%), and plasminogen binding protein (94%).36,37 The serologic marker with greater diagnostic value is IgG4, with an 80% sensitivity and a 98% specificity. Values 2 times greater than normal (140mg/dL) are highly suggestive of autoimmune pancreatitis.19
Treatment decisions are based on data of observational studies. Expectant treatment has been considered, since some patients show spontaneous resolution of pancreatic oedema and narrowing of the pancreatic duct.38 Glucocorticoids are indicated in patients with obstructive jaundice, abdominal or back pain, and extra-pancreatic symptomatic lesions because this has been shown to prevent complications, and control symptoms in up to two thirds of patients.39–41 Rate of remission with steroid therapy has been described to be as high as 98% with an average remission time of 98 days.42 The recommended dose of prednisone is 0.6mg/kg daily for 2 to 4 weeks, and then it should be decreased by 5mg every 1 to 2 weeks depending on the clinical response of the patient, and on the serologic and radiologic evidence of improvement.43 The recommended maintenance dose ranges from 5–10mg daily during the following 2–3 months after the initial schedule is completed.43–45 Suspension of medication after achieving remission has been associated to a greater rate of relapses.46 There is no consensus regarding the exact duration of treatment with steroids, but it is recommended that it should be continued for 3 years for patients that show serologic and radiologic improvement.41 Once the treatment is suspended, the patient needs to be monitored to detect relapses. If there is a relapse, the patient must be started on the same initial dose of prednisone as described above, but the subsequent doses have to be decreased at a more gradual rate.41
Immunomodulatory agents have also been used to control the disease. The most widely used has been azathioprine at a dose of 50mg daily. Others, such as mycophenolate and cyclophosphamide, have also been administered to patients with refractory disease, but there are no clear indications for their use.15,18 There are two case-reports where rituximab was administered with improvement of symptoms in patients with autoimmune pancreatitis refractory to treatment with corticoids.47
ConclusionsAutoimmune pancreatitis is a rare disease that may present with systemic compromise and should be considered as a differential diagnosis of pancreatic tumours. Once there is a clinical suspicion, diagnosis should be made using current diagnostic criteria. When indicated, treatment with steroids must be initiated rapidly in order to improve symptoms and avoid relapses. There is a need for new studies that help determine the ideal duration of treatment as well as the accurate management of relapses and disease refractory to treatment with steroids.
Conflict of InterestsNo conflict of interests reported.
