




Common variable immunodeficiency (CVID) is a rare condition characterized by a constitutional humoral immune deficiency. Its association with systemic lupus is extremely rare.
Case presentationIn this case report, we present a 21-year-old woman with a history of Hashimoto's thyroiditis, who, following the onset of nephrotic syndrome revealing her lupus disease, subsequently developed clinical and laboratory features consistent with CVID, alongside recurrent lupus flares. Her initial therapeutic management was intricate, ultimately resulting in stable remission achieved through monthly immunoglobulin infusions.
ConclusionThis case illustrates the diagnostic and therapeutic difficulty of lupus disease and a common variable immune deficiency.
La inmunodeficiencia común variable es una enfermedad rara, caracterizada por una deficiencia inmunológica humoral constitucional. Su asociación con el lupus eritematoso sistémico es extremadamente infrecuente.
Presentación del casoPresentamos el caso de una mujer de 21 años con antecedentes de tiroiditis de Hashimoto, quien después de desarrollar síndrome nefrótico que reveló la presencia de su enfermedad lúpica, manifestó características clínicas y de laboratorio compatibles con inmunodeficiencia común variable, además de recaídas recurrentes de lupus. El manejo terapéutico inicial fue complejo y finalmente resultó en una remisión estable lograda mediante infusiones mensuales de inmunoglobulina.
ConclusiónEste caso ilustra la dificultad en el diagnóstico y manejo de una enfermedad lúpica y una inmunodeficiencia común variable.
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by a breakdown of immune tolerance to self-antigens, particularly nuclear antigens. This leads to the production of autoantibodies and immune complexes which in turn provoke inflammation and damage in various organs.1 Paradoxically, lupus patients experience immunosuppression, making them more susceptible to infection. This susceptibility is due to several mechanisms, including certain genetic factors, disease-related dysfunction of cellular immune effectors, and the effects of immunosuppressive treatments.1
Rarely, lupus may be associated with common variable immunodeficiency (CVID),1,2 a rare condition associated with a constitutional humoral immune deficiency. Clinically, CVID presents with a diverse range of symptoms and variable hypogammaglobulinemia levels. This exceptional situation can significantly impact the prognosis of lupus patients, presenting both diagnostic challenges and therapeutic challenges.
Case presentationA 21-year-old woman was admitted to the internal medicine department in January 2021 for investigation of a nephrotic syndrome evolving over several weeks. Her medical history included Hashimoto's thyroiditis for the past 6 years under opotherapy (Levothyrox®), and her deceased mother suffered from antiphospholipid syndrome secondary to systemic lupus erythematosus (SLE).
Clinical examination revealed an impure nephrotic syndrome characterized by arterial hypertension associated with a state of anasarca (peregrine edema, edema of the lower limbs, pericardial, pleural and peritoneal effusions), proteinuria at 3g/24h and hypoalbuminemia at 27g/L. The patient also presented with a vespertilio rash, non-scarring alopecia and inflammatory arthralgias. On protein electrophoresis, the gamma globulin level was normal at 15g/L, as were the other globulin fractions. HEp-2 IFA tests revealed the presence of AC-4/5 pattern (speckled Anti-Cell) at 1/640. The semi-quantitative interpretation showed anti-nucleosome antibodies ++, anti-native DNA ++, anti-Sm +++, anti-nRNP: Sm +++, anti-U1-RNP +++, and anti-SSA+. Immunologic tests show Low C3 AND low C4. A renal biopsy confirmed the diagnosis of class IV and V lupus nephropathy according to the International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification revised in 2018.3 This clinical-biological presentation fulfilled 41 points of the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) 2019 classification4 and evoked high disease activity with a SLEDAI score of 24 points.
The patient's treatment regimen included three doses of methylprednisolone at 500mg each, followed by oral prednisone at 1mg/kg per day. This was supplemented by an intravenous bolus of cyclophosphamide at 500mg according to the EUROLUPUS protocol,5 in addition to hydroxychloroquine at a daily dose of 400mg and an angiotensin II receptor antagonist.
However, one week after the start of this treatment, the patient developed sepsis complicated by acute anuric renal failure requiring emergency dialysis. The infectious presentation included an abscessing pyogenic multilobar excavated infectious pneumopathy, bilateral pyelitis, and unilateral right endophthalmitis caused by three pathogens (Staphylococcus, cytomegalovirus, and aspergillosis).
In response to this severe infectious complication, the immunosuppressive therapy was promptly discontinued, and there was a rapid reduction in corticosteroid dosage. Antibiotic therapy was initiated, comprising Imipenem/Cilastatin, ciprofloxacin, valganciclovir, and voriconazole. Managing this situation required three months of hospitalization with daily dialysis sessions until renal function was restored. Although the infection was successfully managed, the endophthalmitis resulted in corneal perforation, requiring evisceration and an ocular prosthesis.
The introduction of an immunosuppressive agent to treat the proliferative nephropathy was a challenge due to the appearance of persistent hypogammaglobulinemia during the second month of hospitalization, suggesting a potentially multifactorial immune deficiency related to the initial nephrotic syndrome and the use of immunosuppressive agents. It was finally decided to treat the patient with mycophenolate mofetil 1g/day, which was discontinued after three weeks due to persistent profound hypogammaglobulinemia of 1.2g/dl.
After three months of remission, the patient was hospitalized for a flare of cutaneous and renal lupus. She presented with progressive skin lesions of epidermal necrolysis type associated with bacterial superinfection in addition to proteinuria at 5g/24h. Local antibiotic treatment was combined with systemic and local corticosteroid therapy, while antiviral therapy (valganciclovir and voriconazole) and hydroxychloroquine were maintained. This episode was concomitant with a mild COVID-19 infection. The clinical–biological evolution was favorable.
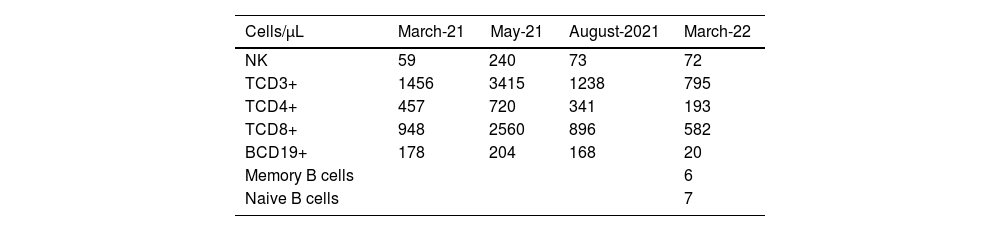
At this stage, the diagnosis of variable common immunodeficiency was retained according to the 2015 revised criteria of the European Society for Immune Deficiencies (ESID).6 Indeed, elements such as persistent pan-hypogammaglobulinemia after cessation of all immunosuppressive treatments and after the disappearance of the nephrotic syndrome, an impairment affecting all lineages with a marked decrease in IgA and IgM, poor vaccine response was demonstrated by post-vaccination serology showing the presence of protective IgG tetanus antitoxin antibodies at low titer, and absence of protective IgG antibody to diphtheria toxoid, and a decrease in switched memory B cells is noted by the reduction of all antibody classes (IgM 0.20g/L, IgG 0.64g/L, IgA 0.43g/L), as well as the collapse of B cell counts (CD19+ B cells 20cells/μL [110–570], memory B cells 7cells/μL [10–31], naive B cells 6cells/μL [92–199]), in the absence of a profound T-cell deficit (CD3+ T cells 795cells/μL [1000–2200], CD8+ T cells 582cells/μL [330–920], CD4+ T cells 193cells/μL [530–1300]) (Table 1), these findings argue for this diagnosis (Table 2).
Diagnostic arguments for CVID in the patient according to the revised ESID 2015 criteria.
| • Evocative situation:- Increased susceptibility to infection- Autoimmune manifestation• Humoral immunity deficiency controlled on more than 2 times:- Marked decrease in IgG (often s 5g/L)- Marked decrease in IgA- Decrease in IgM- Low vaccine response- Decrease in switched memory B cells (LB smB).• Exclusion of causes of secondary hypogammaglobulinemia.• Absence of profound T cell deficiency. |
CVID: common variable immunodeficiency; ESID: European Society of Immune Deficiencies.
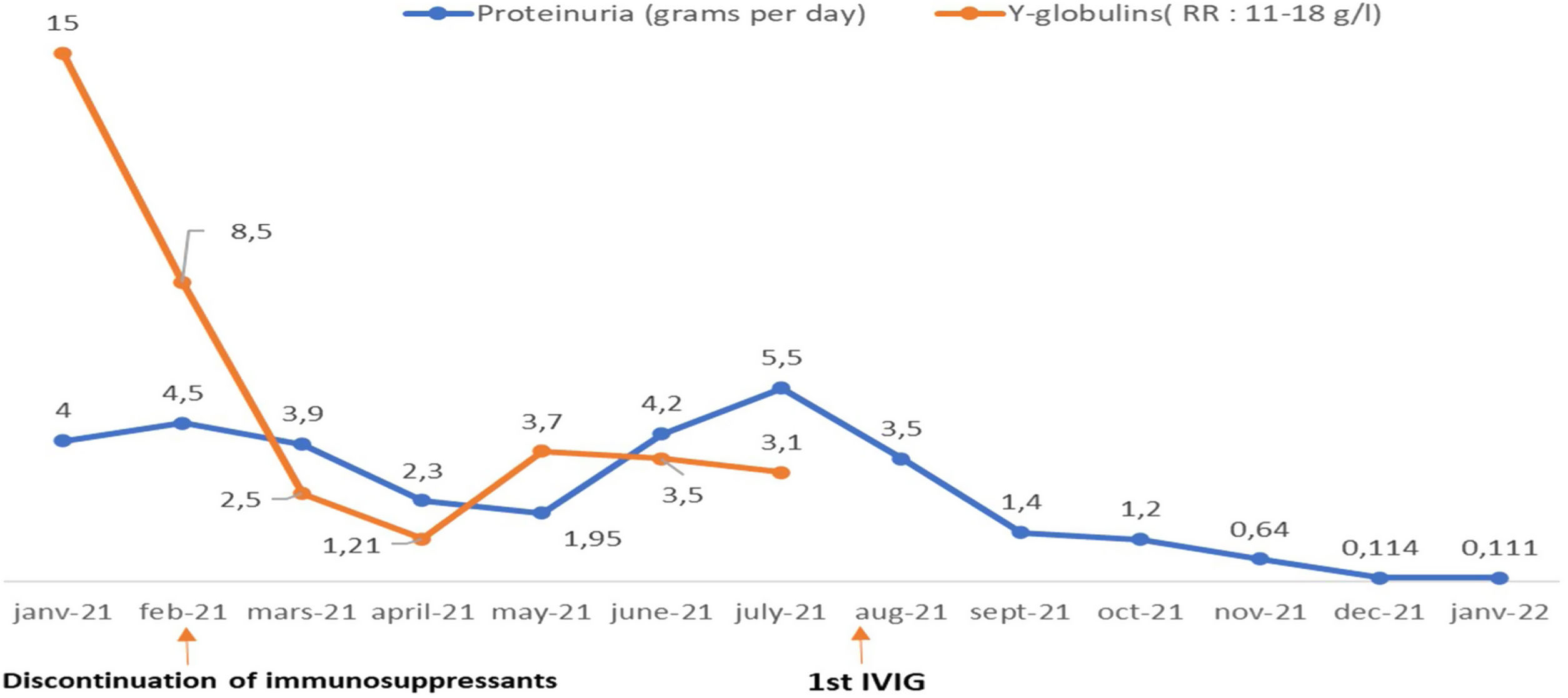
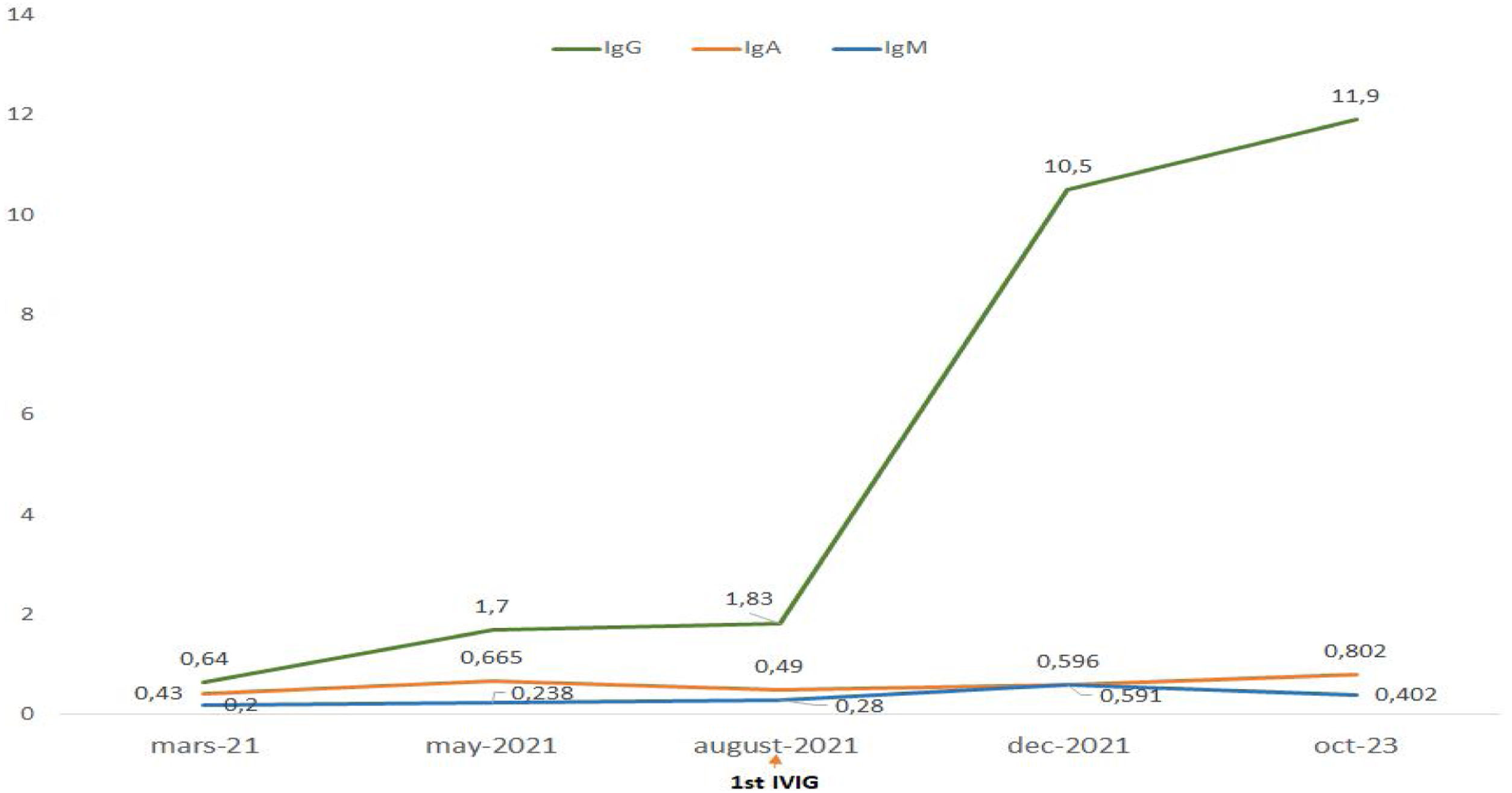
Treatment with intravenous polyvalent immunoglobulin (IVIG) replacement therapy was initiated. Currently, the patient has been in complete remission for two years, following a regimen that includes hydroxychloroquine at a daily dose of 400mg, prednisone at 5mg/day, an angiotensin II receptor antagonist, monthly IVIG infusions, and opotherapy. Fig. 1 shows the temporal changes in proteinuria and gamma globulin levels. Fig. 2 presents the immunoglobulin weight distribution.

 IgA (RR: 0.7–4.3) IgM (RR: 0.5–2.1).")
The association between systemic lupus erythematosus (SLE) and CVID remains extremely rare. Only 15 cases were reported in a recent review.7 Common variable immunodeficiency (CVID) is a heterogeneous group of disorders characterized by hypogammaglobulinemia. It presents in two distinct clinical forms: one is mainly limited to infections, while the other includes a variety of non-infectious, autoimmune, inflammatory, granulomatous and/or lymphoproliferative manifestations. The latter form, which is the most common in internal medicine, often reveals the disease.6
In this association, the diagnosis of SLE most often precedes that of CVID, with an average delay of nine years between the two diagnoses.7 In our case, CVID manifested early, at the same time as SLE. Nevertheless, establishing the diagnosis of this co-occurrence necessitated six months of rigorous investigation and vigilant immune status monitoring after renal remission and the discontinuation of immunosuppressive treatment.
In documented cases, the onset of CVID was generally marked by recurrent infections, mainly of sinonasal or pulmonary origin. Isolated cases of enteritis, urinary tract infection, meningitis and cellulitis have also been reported.7 In our case, the severe clinical presentation of sepsis can be partly attributed to the immunosuppression resulting from the relapse of proliferative lupus nephropathy and the immunosuppressive therapy (high-dose corticosteroids and cyclophosphamide). These two factors have been identified as predictors of infection in several studies.8,9 However, it is important to note that a second clinical presentation without signs of infection is possible. In this context, lupus itself is an expression of CVID, and it is the presence of hypogammaglobulinemia that evokes this association, as noted by Geneviève et al.10 These observations highlight the importance of monitoring immunoglobulin levels in SLE patients. Furthermore, the presence of recurrent infections in such a patient, regardless of SLE activity and/or immunosuppressive treatment, should alert the clinician to the possibility of a potential CVID.
The diagnosis of CVID is currently well codified thanks to the International Diagnostic Criteria (ICON) for CVID, revised in 2016.6 In addition to the need to exclude a secondary cause of hypogammaglobulinemia, the evaluation of CVID must include several elements for a complete characterization of the disease. These include a quantitative immunoglobulin test, an assessment of the vaccine response, a detailed analysis of the B and T lymphocyte phenotype, and eventually a search for mutations associated with severe combined immunodeficiency.6,11 Although these criteria have high sensitivity and specificity, they remain difficult to perform, especially in developing countries or in centers without immunology laboratories. In our opinion, this situation may underestimate the frequency of CVID in SLE patients.
The association of lupus with CVID presents a significant treatment challenge. In our patient's case, the situation was particularly complex. Despite the severity of the renal impairment, the uncontrollable infectious state led to the discontinuation of immunosuppressive therapy. The subsequent decision to reintroduce it proved difficult given the increased risk of infection. This problem was solved by the maintenance of renal remission after the introduction of intravenous immunoglobulin (IVIG) substitution with low-dose corticosteroids and a synthetic antimalarial.
The favorable evolution of SLE after the development of CVID has been documented and explained by the hypothesis that if a patient with SLE is no longer able to secrete antibodies, it would be conceivable that aspects of the disease generally attributed to the production of pathogenic autoantibodies might improve.7,12 However, contrary to expectations, this trend is not systematic. In fact, cases of unfavorable course of lupus have been reported.13 This raises important questions about the long-term management of patients with these two comorbidities. Finding the right balance between controlling autoimmunity and preserving the immune response against infection is a complex challenge.
ConclusionThe association between systemic lupus erythematosus (SLE) and common variable immunodeficiency (CVID) is rare and often poorly known. It is important to make physicians aware of the need for clinical surveillance for infectious warning signs and regular monitoring of gamma globulin levels in patients with SLE. This would help to better identify and manage this association. Further research is needed to better understand the mechanisms underlying this association and to develop more effective therapeutic strategies.
Ethical considerationsThe patient signed and approved the informed consent for publication of the clinical case report. The authors state that this article does not contain any personally identifiable information about patients.
FinancingNone.
Conflicts of interestNone.
