



El síndrome de encefalopatía posterior reversible (PRES) es un trastorno clínico-radiológico caracterizado por encefalopatía, convulsiones, cefalea y alteraciones visuales. Se asocia a entidades que ocasionan daño endotelial, causando edema cerebral vasogénico evidente en resonancia magnética. En pacientes con lupus eritematoso sistémico (LES) se presenta en <1%. Con tratamiento oportuno usualmente resuelve; caso contrario puede producir secuelas neurológicas o muerte. Se reporta el caso de PRES en una paciente con LES con actividad severa, emergencia hipertensiva y glomerulonefritis lúpica que comienza con estatus epiléptico. Evolucionó satisfactoriamente con tratamiento anticonvulsivante, antiedema cerebral, antihipertensivo y control de los demás factores causales.
Posterior reversible encephalopathy syndrome (PRES) is a clinical-radiological disorder that may include encephalopathy, seizures, headache, and visual disturbances. It is associated with conditions that induce endothelial damage, causing vasogenic cerebral oedema that can be observed in magnetic resonance scans. It occurs in <1% of patients with systemic lupus erythematosus (SLE). It is usually resolved with timely treatment, but delays may lead to neurological sequelae or death. A case of PRES is presented in a patient with SLE with severe activity, a hypertensive emergency, and lupus glomerulonephritis debuting with epileptic status. The outcome was satisfactory with anticonvulsants, as well as treatment for her cerebral oedema and hypertension, along with control of other causal factors.
El síndrome de encefalopatía posterior reversible (PRES) es una rara complicación de diversas entidades clínicas. Su incidencia es desconocida, habiéndose reportado en un rango amplio de edad de 14 a 78 años, con una edad media de 44 años y una proporción varón/mujer de 0,8/11. Aunque el pronóstico suele ser favorable, se han reportado tasas de mortalidad de hasta el 15%2. Está determinado por manifestaciones clínico-radiológicas típicas, usualmente transitorias3. De manera aguda o subaguda, en orden descendente de frecuencia se presenta con encefalopatía, convulsiones, cefalea, alteraciones visuales y déficit neurológico focal4. Se han postulado varias teorías fisiopatológicas, siendo 2 las más aceptadas. La primera sugiere que el incremento súbito de la presión arterial supera la autorregulación del flujo sanguíneo cerebral, causando vasodilatación e hiperperfusión, con rotura de la barrera hematoencefálica (BHE) y edema vasogénico5. Así, clásicamente se ha asociado a eclampsia y encefalopatía hipertensiva; sin embargo, del 20 al 30% de los pacientes son normotensos, sugiriendo una segunda teoría de toxicidad endotelial directa causada por mediadores inflamatorios, más correlacionada a pacientes con tratamiento inmunosupresor, fallo renal, conectivopatías o sepsis6. En pacientes con lupus eritematoso sistémico (LES) se presenta en <1%, teniendo mayor incidencia en jóvenes, con Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) ≥6 y comorbilidades asociadas7. La resonancia magnética nuclear (RMN) de cerebro es determinante para el diagnóstico, mostrando edema vasogénico, usualmente en territorio cerebral posterior, bilateral y simétrico8. Dado que el diagnóstico deL PRES requiere alta sospecha clínica y de imagen; con la subsecuente instauración de tratamiento temprano para un pronóstico favorable; el objetivo del presente trabajo es aportar información para el reconocimiento y manejo de este inusual síndrome asociado al LES.
Descripción del casoMujer de 25 años, mestiza, procedente de Quito, Ecuador, con antecedentes patológicos personales de hipotiroidismo y LES, diagnosticados en diciembre del 2012, a los 21 años de edad. En diciembre del 2016, 4 años después del diagnóstico del LES, presenta exacerbación desencadenada por enfermedad diarreica aguda; con manifestaciones músculo-esqueléticas (artritis, mialgias), mucocutáneas (úlceras orales), serositis (derrame pleural derecho), bicitopenia (anemia, trombocitopenia), compromiso renal (hematuria, proteinuria, insuficiencia renal aguda, clasificación Acute Kidney Injury Network [AKIN] III) e hipertensión arterial (HTA); todo lo cual le confirió un SLEDAI elevado (valor: 23). La biopsia renal reportó glomerulonefritis proliferativa focal lúpica clase II, que no es correspondiente con actividad lúpica intensa; sin cambios atribuibles a síndrome antifosfolípido, así como negatividad de estos anticuerpos. Por la afectación multiorgánica recibió pulsos de metilprednisolona de 1g intravenoso por 3 días, reposición de hemoderivados, plasmaféresis 6 sesiones, hemodiálisis, amlodipino 10mg/día, atenolol 50mg/día y micofenolato mofetilo 1g/12h vía oral, ya que presentó intolerancia gastrointestinal a dosis superiores. Tres semanas después de su ingreso presentó mejoría clínica y analítica aceptable al tratamiento, por lo cual se indicó su alta hospitalaria. Veinticuatro horas después reingresa en estatus convulsivo. En emergencias inician manejo de vía aérea, anticonvulsivantes intravenosos con diazepam 10mg, midazolam 3mg, fenitoína 1g y transfieren a la unidad de cuidados intensivos.
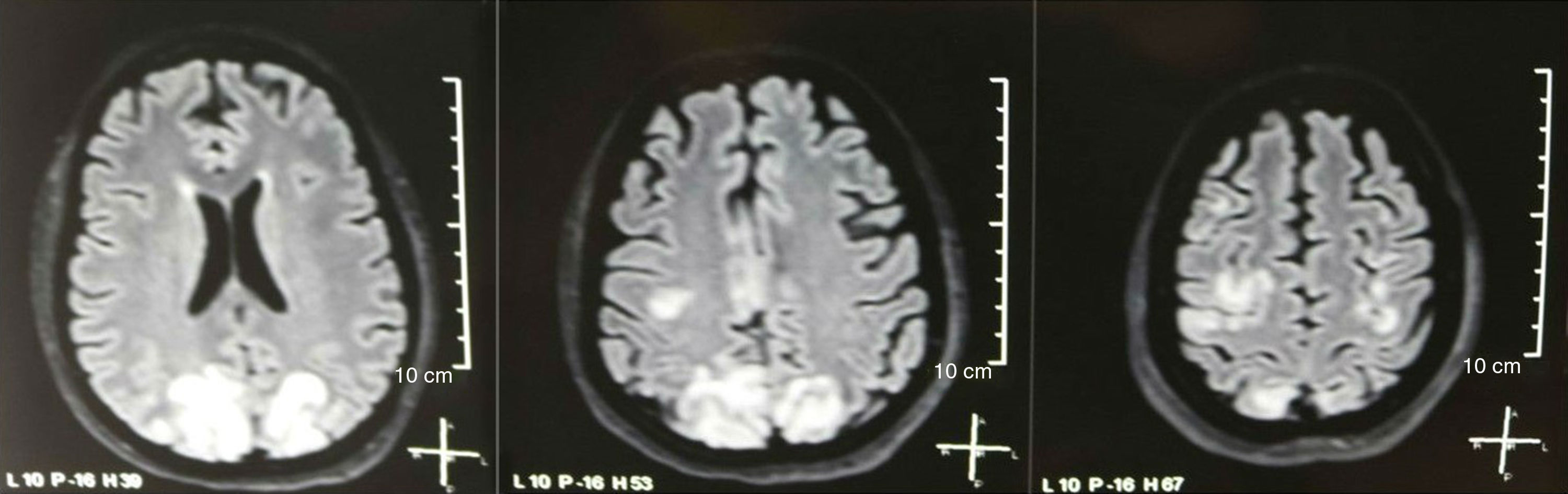
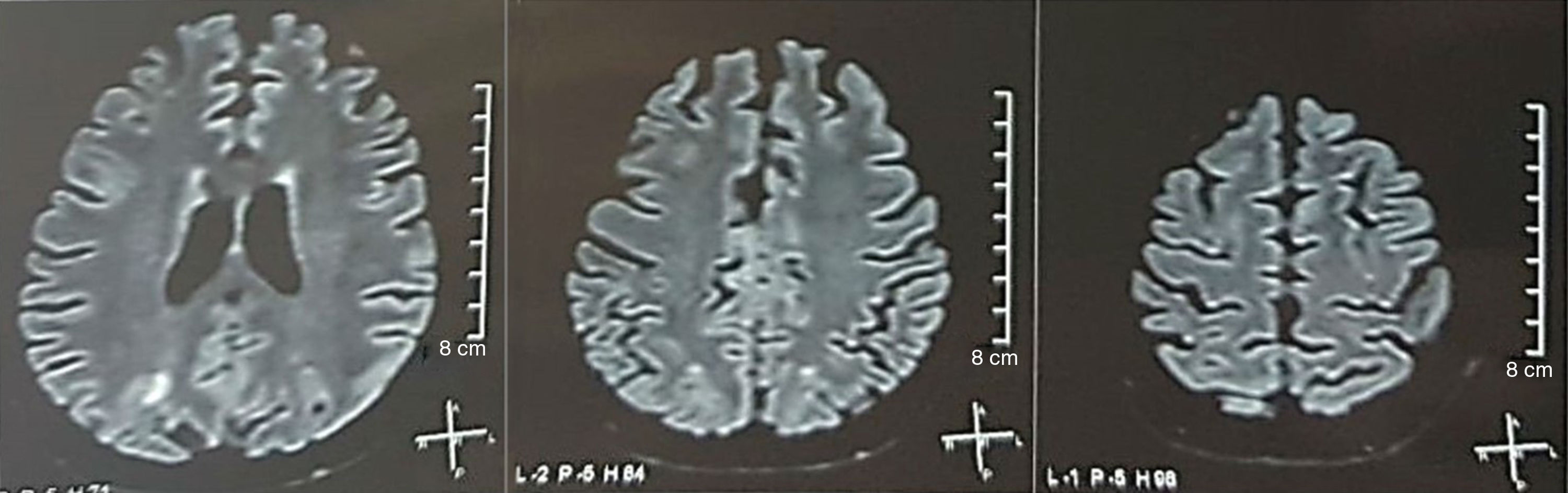
Se solicitó una tomografía de cráneo la cual no mostró signos de isquemia o sangrado; se observó una hipodensidad occipital derecha sin efecto de masa por lo que se amplió la orden de exámenes para filiar su etiología. Se excluyeron causas metabólicas, infecciosas y medicamentosas. Por HTA de difícil control (hasta 190/100mmHg con tensión arterial media [TAM] de 130mmHg), requirió hasta 6 antihipertensivos; por sonda nasogástrica con atenolol 50mg/12h, losartán 100mg/día, amlodipino 10mg/día, doxazosina 2mg/6h e intravenosos con nitroprusiato 50mg/día y furosemida 20mg/6h. Por actividad lúpica severa (SLEDAI 21: convulsiones, hematuria, proteinuria, hipocomplementemia, anti-DNA, trombocitopenia), recibió nuevamente tratamiento con metilprednisolona 1g/3 días. El electroencefalograma no mostró actividad epileptiforme. Se solicitó angiorresonancia cerebral en la cual no se encontraron hallazgos consistentes con vasculitis ni trombosis del sistema nervioso central. En la RMN cerebral se observan imágenes típicas de PRES (fig. 1), cuyo desarrollo estaría con relación al LES exacerbado, HTA severa, glomerulonefritis lúpica y uso de inmunosupresores, por lo que se añadió nimodipino oral 60mg/6h y se controlaron los factores desencadenantes. Por riesgo de lupus inducido por fármacos se retiró gradualmente la fenitoína, con aumento progresivo de levetiracetam hasta 1g/12h por sonda nasogástrica. La tabla 1 detalla los estudios complementarios relevantes. Durante el seguimiento no presentó nuevos eventos convulsivos, la función renal se mantuvo estacionaria, mejoraron las cifras tensionales (TAM 85-90mmHg), disminuyó la actividad lúpica (SLEDAI 13: hematuria, proteinuria, hipocomplementemia, anti-DNA, trombocitopenia). La RMN cerebral de control evidenció involución de las lesiones previas (fig. 2).

Estudios complementarios relevantes del caso
| Fecha | Analíticas |
|---|---|
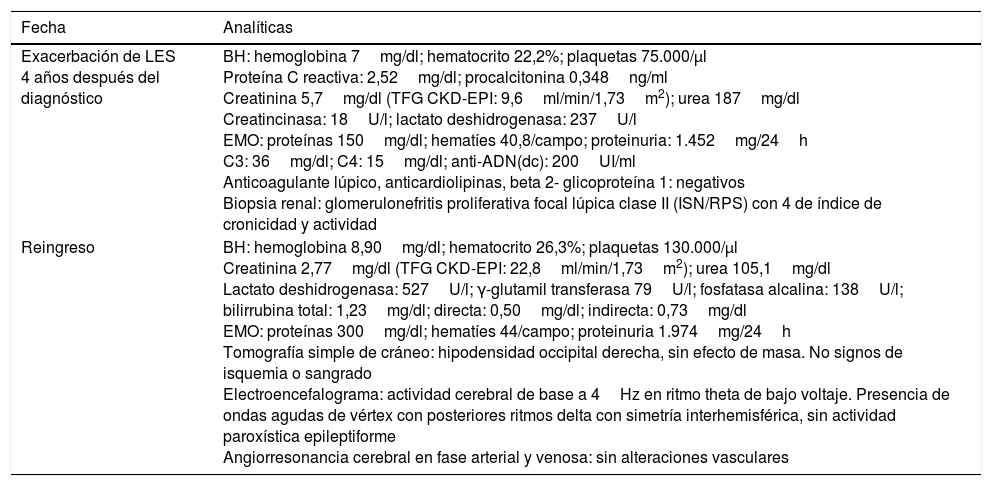
| Exacerbación de LES 4 años después del diagnóstico | BH: hemoglobina 7mg/dl; hematocrito 22,2%; plaquetas 75.000/μl Proteína C reactiva: 2,52mg/dl; procalcitonina 0,348ng/ml Creatinina 5,7mg/dl (TFG CKD-EPI: 9,6ml/min/1,73m2); urea 187mg/dl Creatincinasa: 18U/l; lactato deshidrogenasa: 237U/l EMO: proteínas 150mg/dl; hematíes 40,8/campo; proteinuria: 1.452mg/24h C3: 36mg/dl; C4: 15mg/dl; anti-ADN(dc): 200UI/ml Anticoagulante lúpico, anticardiolipinas, beta 2- glicoproteína 1: negativos Biopsia renal: glomerulonefritis proliferativa focal lúpica clase II (ISN/RPS) con 4 de índice de cronicidad y actividad |
| Reingreso | BH: hemoglobina 8,90mg/dl; hematocrito 26,3%; plaquetas 130.000/μl Creatinina 2,77mg/dl (TFG CKD-EPI: 22,8ml/min/1,73m2); urea 105,1mg/dl Lactato deshidrogenasa: 527U/l; γ-glutamil transferasa 79U/l; fosfatasa alcalina: 138U/l; bilirrubina total: 1,23mg/dl; directa: 0,50mg/dl; indirecta: 0,73mg/dl EMO: proteínas 300mg/dl; hematíes 44/campo; proteinuria 1.974mg/24h Tomografía simple de cráneo: hipodensidad occipital derecha, sin efecto de masa. No signos de isquemia o sangrado Electroencefalograma: actividad cerebral de base a 4Hz en ritmo theta de bajo voltaje. Presencia de ondas agudas de vértex con posteriores ritmos delta con simetría interhemisférica, sin actividad paroxística epileptiforme Angiorresonancia cerebral en fase arterial y venosa: sin alteraciones vasculares |
Anti-ADN(dc): anti-ADN de doble cadena; BH: biometría hemática; EMO: examen microscópico de orina; ISN/RPS: International Society of Nephrology/Renal Pathology Society; LES: lupus eritematoso sistémico; TFG CKD-EPI: tasa de filtrado glomerular estimada por la ecuación del Chronic Kidney Disease-Epidemiology Collaboration.
: lesiones previas en resolución.")
Desde la primera descripción del PRES, realizada en 1996 por Hinchey et al., se ha ampliado el conocimiento de varios aspectos de esta entidad. Su denominación original de síndrome de leucoencefalopatía posterior reversible resultó inapropiada, ya que los cambios imagenológicos no siempre se limitan a la sustancia blanca cerebral y sus manifestaciones clínicas no siempre son reversibles9. Los primeros 15 casos reportados ocurrieron en pacientes con encefalopatía hipertensiva, eclampsia o en tratamiento inmunosupresor10. También se ha observado como complicación de otras entidades como sepsis, fallo renal y conectivopatías; por lo que actualmente se conoce que los factores de riesgo que originan disfunción endotelial son clave para el desarrollo del PRES11. La incidencia global se desconoce, pero los datos de estudios retrospectivos señalan que es más frecuente en personas entre 39-47 años, generalmente mujeres, con comorbilidades como trastornos hipertensivos, renales o autoinmunes12. En pacientes con LES, muchos autoanticuerpos se dirigen contra el endotelio; produciendo su activación, expresión de moléculas de adhesión (E-selectina, VCAM-1, ICAM-1) y exposición a citoquinas proinflamatorias como IL-1β, TNFα e IL-6, causando disrupción de la BHE y aparición de complicaciones neurológicas13. Se ha reportado que en personas diagnosticadas con LES, el PRES se presenta en el contexto de actividad lúpica moderada a severa, así como asociado a insuficiencia renal e hipertensión mal controlada14.
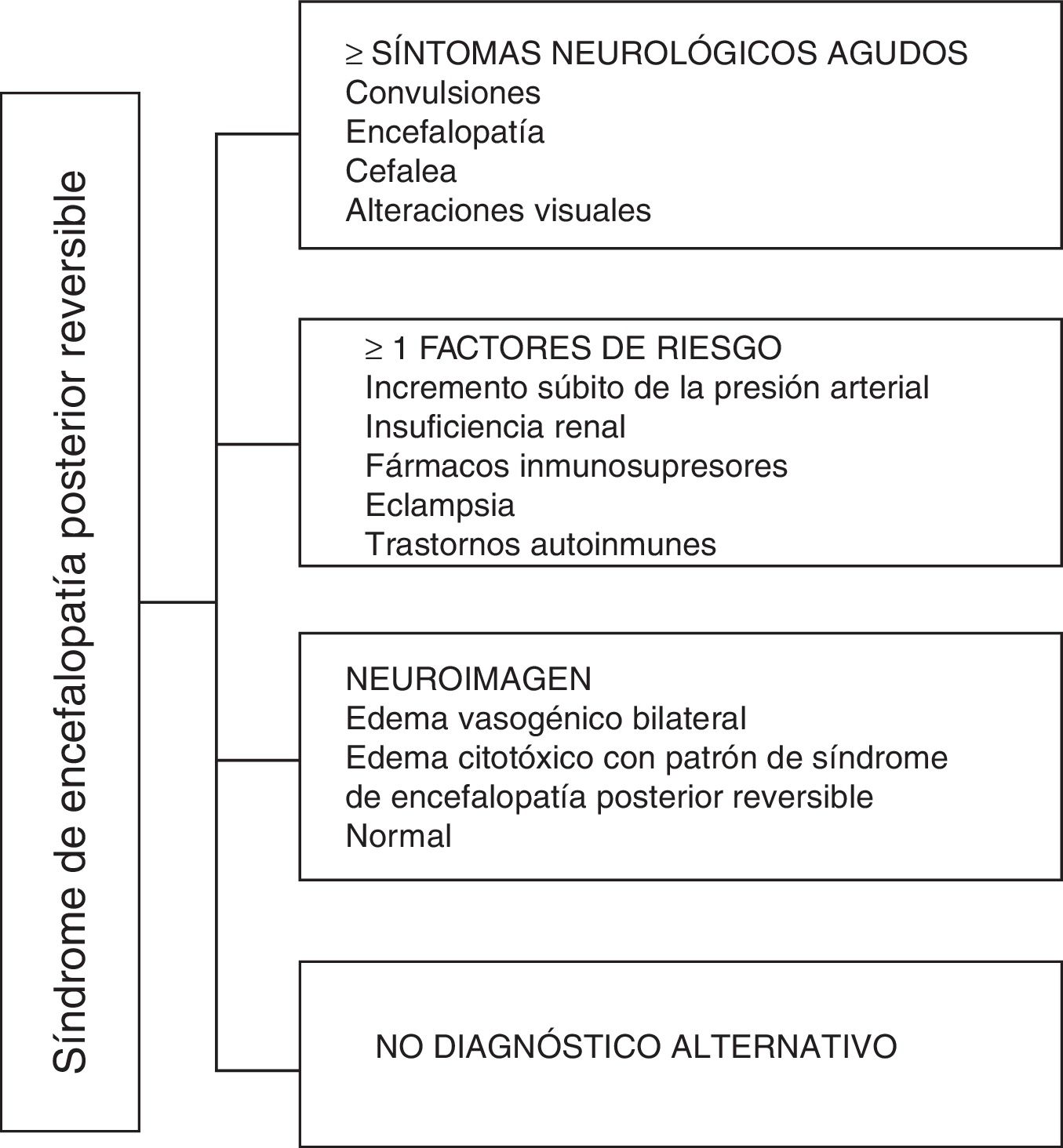
En cuanto a las manifestaciones clínicas del PRES, se caracteriza por grados variables de encefalopatía desde confusión a estupor (50-80%), convulsiones (60-75%), cefalea (50%) y alteraciones visuales que van desde visión borrosa a ceguera cortical (33%); siendo inusual el déficit neurológico focal (10-15%) y el estatus epiléptico (5-15%)15. Para la valoración inicial del compromiso neurológico en estos pacientes suele solicitarse inicialmente una tomografía axial computarizada (TAC) de cráneo, la cual a menudo es normal o puede mostrar hipodensidades córtico-subcorticales, de predominio en regiones cerebrales posteriores16. La RMN de cerebro determina el diagnóstico, mostrando edema vasogénico, usualmente en la sustancia blanca de los lóbulos occipital y parietal (territorio de la circulación cerebral posterior), visualizado como lesiones hipertensas en T2 y FLAIR, bilateral y simétrico17. La afectación preferente de la sustancia blanca se debe a su estructura de fibras mielinizadas, arteriolas y capilares que le confieren una mayor laxitud. De igual manera, los vasos de la circulación cerebral anterior, al poseer mayor innervación simpática pueden responder adecuadamente mediante vasoconstricción ante el aumento súbito del flujo sanguíneo cerebral secundario a hipertensión; mecanismo protector menos desarrollado en el sistema vértebro basilar18. Con menor frecuencia puede afectarse la sustancia gris y otros lóbulos. Las imágenes con secuencias de difusión permiten distinguir entre el edema vasogénico, típico de PRES, del edema citotóxico que puede presentarse de manera atípica y que puede progresar a infarto19. El electroencefalograma no siempre se correlaciona con la afectación neurológica, pero puede revelar encefalopatía por la presencia de ondas focales agudas. En pacientes con convulsiones asociadas a PRES, la principal alteración electroencefalográfica es el enlentecimiento generalizado en frecuencia theta/delta20. El análisis del líquido cefalorraquídeo muestra cambios inespecíficos como aumento leve de la celularidad y proteínas, por lo cual resulta útil cuando convenga descartarse infección del sistema nervioso central21. Adicional a las pruebas mencionadas, deben realizarse las que se consideren necesarias para el diagnóstico diferencial principalmente con neurolupus, encefalopatía metabólica y parainfecciosa, encefalitis, infarto de la arteria cerebral posterior y trastornos desmielinizantes22,23. Con base en lo anterior, en la figura 3 se indica el algoritmo propuesto por Fugate et al. para el diagnóstico de PRES, el cual pretende identificar incluso casos atípicos15.

En nuestra paciente, dada la presentación clínica junto a los múltiples factores de riesgo, hallazgos imagenológicos y exclusión de otras etiologías, se concluyó el diagnóstico del PRES. Se instauró oportunamente tratamiento sintomático con medicación anticonvulsivante y antiedema cerebral, junto al control de los factores causales: HTA severa, LES con actividad severa, glomerulonefritis lúpica, fármacos inmunosupresores; ratificando el diagnóstico durante el seguimiento con la resolución de las alteraciones clínicas e imagenológicas. Con respecto al manejo del PRES se debe reducir la presión arterial, tratar las convulsiones y controlar el desencadenante. La disminución rápida de la presión arterial podría provocar isquemia cerebral, por lo que se sugiere un objetivo de presión arterial media entre 105-125mmHg, sin sobrepasar el 25% de esta reducción en la primera hora. Los fármacos de primera línea son los bloqueadores de los canales de calcio (nicardipina o de elección nimodipino que además previene el vasoespasmo cerebral) o bloqueadores beta (por ejemplo, labetalol). Como segunda línea puede utilizarse nitroprusiato sódico o hidralazina. Debe evitarse la nitroglicerina por su efecto vasodilatador, que aumentaría el edema cerebral24. El tratamiento de las convulsiones es semejante al de otras convulsiones epilépticas. De primera línea se usan benzodiacepinas como lorazepam o diazepam. De segunda línea, fenitoína o valproato sobre todo en estatus epiléptico o fenobarbital. En embarazadas se puede utilizar sulfato de magnesio. En convulsiones refractarias pueden administrarse propofol o pentobarbital25. En pacientes con LES se procura evitar medicamentos que podrían causar lupus inducido por fármacos como hidralazina, metildopa, captopril, fenitoína, valproato y carbamazepina. Existe controversia sobre el manejo de fármacos inmunosupresores en el tratamiento del PRES en pacientes con LES26. Después de la resolución del PRES, las convulsiones son infrecuentes, por lo que se debe considerar suspender los anticonvulsivantes siempre que exista control adecuado de los factores de riesgo27. Con tratamiento oportuno y adecuado, la mayoría de los pacientes con PRES evoluciona satisfactoriamente con remisión de los síntomas y de las lesiones imagenológicas en días a semanas, aunque se han observado complicaciones sobre todo hemorrágicas entre el 9-33% de los casos, por lo que este caso resalta la importancia de su reconocimiento y manejo, que a menudo resulta todo un reto28.
ConclusionesEl diagnóstico del PRES requiere alta sospecha clínica y de imagen. El tratamiento oportuno con control de los síntomas y de la causa subyacente ratifica el diagnóstico durante el seguimiento, con la resolución de las alteraciones clínicas e imagenológicas; caso contrario puede ocasionar secuelas neurológicas o muerte.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos al personal de los servicios de neurología, nefrología y a la unidad de cuidados intensivos de nuestra institución, quienes contribuyeron al manejo de la paciente; agradecemos también a la paciente y a sus familiares por su colaboración y confianza.
