





La enfermedad de Behçet es una enfermedad inflamatoria sistémica que afecta principalmente a la mucosa oral y genital, la piel y los ojos. Se llevó a cabo una revisión sistemática cualitativa de la literatura en búsqueda de encontrar la epidemiología, la etiopatogenia, el espectro de manifestaciones clínicas, el diagnóstico, el enfoque de tratamiento y los clinical trials actuales de esta enfermedad en la población pediátrica. Se hizo una búsqueda bibliográfica en PubMed sin restricción de idioma ni fecha de publicación. Se examinaron las referencias de los artículos incluidos en busca de literatura relevante adicional. La búsqueda inicial arrojó un total de 570 estudios provenientes de PubMed, uno de página web, 4 del Ministerio de Salud colombiano y 4 de revisión de citas bibliográficas, de los cuales se incluyeron 32 artículos para la presente revisión, y se encontró que la prevalencia a nivel global se calcula en alrededor de 10,3 por 100.000 habitantes. La enfermedad de Behçet es una vasculitis que afecta a los vasos de todos los tamaños, incluyendo las venas. Se reclasificó recientemente como vasculitis de tipo variable. Otras manifestaciones clínicas frecuentes son las articulares, las cutáneas y las digestivas. Aunque su etiopatogenia no es clara, en los últimos años se ha considerado que se trata de una entidad autoinflamatoria multicausal. Su diagnóstico es principalmente clínico. El manejo debe individualizarse en función de las manifestaciones de la enfermedad, dada la variabilidad clínica. Se necesitan estudios multicéntricos, controlados con placebo y estandarizados que involucren grandes series de pacientes, utilicen puntuaciones clínicas y tengan un seguimiento a largo plazo para comprender mejor la naturaleza de esta enfermedad.
Behçet's disease is a systemic inflammatory disease that mainly affects the oral and genital mucosa, skin and eyes. A qualitative systematic review of the literature is carried out in search of finding the epidemiology, etiopathogenesis, spectrum of clinical manifestations, diagnosis, treatment approach and current Clinical Trials of Behçet's disease in the pediatric population. A bibliographic search was performed in PubMed without language or publication date restrictions. References of included articles were examined for additional relevant literature. The initial search yielded a total of 570 studies from PubMed, one from a website, 4 from the Colombian Ministry of Health and 4 from a review of bibliographic citations, of which 32 articles were included for the present review, finding that the prevalence at a global level it is estimated around 10.3 per 100,000 inhabitants. Behçet's disease is a vasculitis that affects vessels of all sizes including veins. It was recently reclassified as variable type vasculitis. Other frequent clinical manifestations are joint, skin and digestive manifestations. Although its etiopathogenesis is not clear, in recent years it has been considered a multicausal autoinflammatory entity. Its diagnosis is mainly clinical. Management should be individualized based on the manifestations of the disease given the clinical variability. Multicenter, placebo-controlled, standardized studies that involve large series of patients, use clinical scores, and have long-term follow-up are needed to better understand the nature of this disease.
La enfermedad de Behçet (EB) es una enfermedad inflamatoria sistémica que afecta principalmente a la mucosa oral y genital, la piel y los ojos. Es una vasculitis que afecta a los vasos de cualquier tamaño, incluyendo las venas. Se reclasificó recientemente como vasculitis de tipo variable1.
La distribución geográfica de la EB se relaciona estrecha e históricamente con la ruta de la seda. En 1937 Hulusi Behçet la definió como una enfermedad que cursa con aftas orales y genitales recurrentes, con uveítis. La concepción actual es de una vasculitis sistémica que puede afectar a vasos sanguíneos de cualquier tamaño y tipo e involucrar a casi cualquier sistema, incluido el gastrointestinal, el nervioso, el musculoesquelético y el cardiovascular. La enfermedad generalmente se desarrolla en la edad adulta joven, entre la segunda y la cuarta década de la vida. Alrededor del 15-20% de todos los casos son de inicio en la infancia. La EB pediátrica difiere de la EB en adultos no solo en la edad de inicio, sino también en la frecuencia y la distribución de los hallazgos clínicos, la gravedad de la enfermedad y el pronóstico1–3.
Como se observa en las enfermedades autoinflamatorias, las recaídas de EB son recurrentes y autolimitadas. La afectación neurológica y ocular ocasiona importantes limitaciones funcionales. La vasculitis de grandes vasos es la principal causa de muerte, típicamente por trombosis múltiple o arteritis pulmonar. Aunque se considera una sola entidad, la presentación clínica de la EB es heterogénea y puede variar según el sexo, el país de residencia y la edad de aparición. Los mecanismos subyacentes que causan la enfermedad aún no están claros y podrían combinar tanto la autoinflamación como la autoinmunidad3–7.
Se hizo una revisión sistemática cualitativa de la literatura a fin de responder las siguientes preguntas relacionadas con la EB en la población pediátrica:
- 1.
¿Cuál es la epidemiología global y local de esta enfermedad?
- 2.
¿Qué se sabe de su etiopatogenia?
- 3.
¿Cuál es el espectro de manifestaciones clínicas?
- 4.
¿Cuál es el enfoque diagnóstico de esta dolencia?
- 5.
¿Cuál es el enfoque de tratamiento y cuáles son los clinical trials actuales para esta enfermedad?
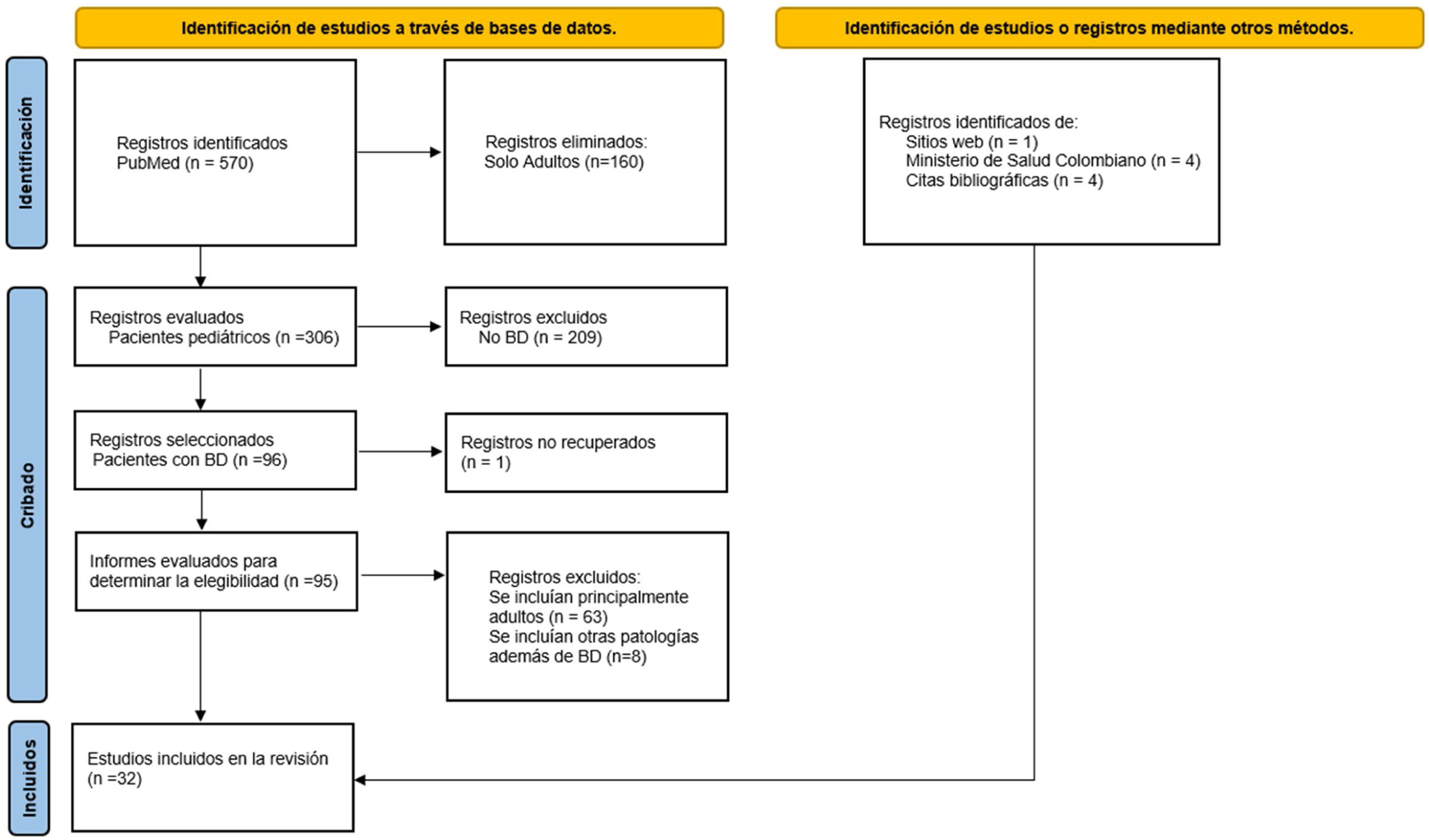
Se hizo una búsqueda bibliográfica en PubMed sin restricción de idioma ni fecha de publicación. Se examinaron las referencias de los artículos incluidos en busca de literatura relevante adicional. Se utilizaron los términos de búsqueda relacionados: «Behçet disease», «Pediatric OR Child». En la construcción de las ecuaciones de búsqueda, cada uno de los términos MeSH se cruzó con las respectivas palabras clave, utilizando diferentes operadores booleanos (OR, AND). Se siguió la extensión de la guía PRISMA para revisiones panorámicas (en inglés, extension for scoping reviews) (fig. 1).

De conformidad con la normativa colombiana (Resolución 008430 de 1993 del Ministerio de Salud), esta revisión sistemática se considera una investigación sin riesgo, debido a que no se hizo ninguna intervención o modificación intencionada de las variables biológicas, fisiológicas, psicológicas o sociales de las personas.
ResultadosLa búsqueda inicial arrojó un total de 570 estudios provenientes de PubMed. Luego de excluir artículos que solo mencionaban adultos, se llevó a cabo el cribado y la elegibilidad por medio de la revisión de los abstracts y de los textos completos que cumplieran con las respuestas a las preguntas objeto del estudio (fig. 1). Los resultados se exponen de manera detallada a continuación.
De los 32 artículos finalmente incluidos, 15 eran revisión de tema, una carta al editor, 2 reportes de caso, 3 series de casos, un estudio descriptivo observacional, 2 estudios observacionales basados en registro, 2 estudios de cohortes, un estudio de casos y controles, una página web y 4 del Ministerio de Salud colombiano.
EpidemiologíaLa prevalencia de EB a nivel global es de alrededor de 10,3 por 100.000 habitantes, siendo Jordania (664/100.000 habitantes) y Turquía (600/100.000 habitantes) los países de mayor reporte, mientras que Escocia (0,3/100.000 habitantes) es el de notificación más baja3. El 4-26% de los pacientes con EB tiene un inicio pediátrico definido como la aparición de síntomas antes de los 16 años. La EB adulta es de inicio insidioso, con su pico entre los 25-30 años3–6.
La edad media de aparición de la EB pediátrica varía de 4,9 a 12,3 años y el retraso en el diagnóstico es de unos 3 años en las cohortes pediátricas notificadas; los hombres tienen un curso de la enfermedad más grave que las mujeres3,6.
En el 2020 se estableció un marco colaborativo internacional para la investigación sobre enfermedades autoinflamatorias y enfermedades oculares inmunomediadas, con más de 170 centros de atención clínica en todo el mundo, para disminuir el subregistro y la falta de información de adherencia al manejo por parte de esta población1,8. En Colombia, se considera que es una enfermedad huérfana, de notificación obligatoria para el Instituto Nacional de Salud, según la Resolución 946 de 2019, la Resolución 023 de 2023 y el Código 342 de 20229,10. En el año 2019 Colombia reportó 75 casos, sin información de la edad de aparición11.
EtiopatogeniaLa etiopatogenia de la EB aún no ha podido esclarecerse por completo. La opinión más ampliamente aceptada es que varios desencadenantes infecciosos o ambientales desempeñan un papel en individuos con susceptibilidad genética. Se han investigado en la etiología de la EB bacterias como Streptococcus sanguis, Helicobacter pylori y Mycoplasma, e infecciones virales, incluidos el virus del herpes simple 1, el virus de Epstein-Barr, los virus de la hepatitis y el citomegalovirus3,12,13.
Se han identificado Staphylococcus aureus y Streptococcus oralis en las lesiones cutáneas de pacientes con esta enfermedad. Los estudios evidencian que los autoantígenos, a través del mimetismo molecular, desempeñan un papel clave en su desarrollo. En este sentido, se han observado varios autoantígenos, incluidas las proteínas de choque térmico, el antígeno S, la proteína de unión a retinoides interfotorreceptores, la α-tropomiosina y la αβ-cristalina. Mediante el estudio de la homología de secuencia entre las proteínas M de la pared celular estreptocócica y la tropomiosina, se revelaron epítopos inmunológicos compartidos. La similitud entre esta proteína de superficie estreptocócica y la tropomiosina muestra que el mimetismo molecular puede conducir a la inflamación observada en la EB, por medio de la inducción de una inmunorreacción a la tropomiosina12,13.
Ahora bien, se cree que la enfermedad tiene mecanismos patogénicos que se asemejan a enfermedades autoinmunes, autoinflamatorias y espondiloartropatías seronegativas. Uno de los temas más discutidos es el de los componentes genéticos de la enfermedad. El antígeno leucocitario humano (HLA) B51 es el factor genético predisponente más conocido para la EB y su positividad aumenta el riesgo de desarrollarla en 5,78 veces3,6,12,13.
La homocigosidad para la mutación que codifica ERAP1 p.Arg725Gln se asoció con un mayor índice de probabilidades de EB en individuos HLA-B51+, en comparación con individuos HLA-B51−. ERAP1 es una aminopeptidasa expresada en el retículo endoplásmico que mejora o limita los péptidos antigénicos para optimizar el tamaño antes de cargarlos en moléculas del MHC de clase i, como HLA-B51. Estas alteraciones en el peptidoma pueden influir negativamente en la presentación de antígenos de las moléculas del MHC-I. Se plantea la hipótesis de que los peptidomas de menor afinidad pueden inducir una respuesta de células T tolerante o autoinmune específica. Los cambios en la afinidad por el peptidoma afectan la cinética de plegamiento de la molécula MHC-I. De manera comparable, este plegamiento incorrecto de las moléculas de MHC-I también se observa en HLA-B27 y se propone como uno de los mecanismos por los cuales la EB se define como una enfermedad de MHC-I13.
Las células natural killer son uno de los componentes principales de la inmunidad innata. En las enfermedades autoinmunes, su frecuencia disminuye y su citotoxicidad se ve afectada, lo que evidencia que estas células desempeñan un papel protector en el control de la autoinmunidad12. Hasan et al. informaron que la cantidad de células natural killer en la sangre periférica de pacientes con EB disminuyó significativamente, un cambio que estaba relacionado con la actividad de la enfermedad. Además, ese estudio reveló que las células natural killer CD56bright CD16− y CD56dim CD16+ se agotaron en la sangre periférica de pacientes con EB en comparación con la cantidad de estas células en controles sanos13,14, lo que resulta en un aumento de estas células citotóxicas en los sitios inflamatorios y la activación y el mantenimiento de la inflamación del tejido en pacientes con EB a través de la producción de citocinas Th1, lo que resulta en citotoxicidad en la fase activa12–14.
Se supone que un sistema inmunológico adaptativo desregulado con un aumento de citocinas proinflamatorias contribuye a los síntomas inflamatorios de los pacientes con EB. El sistema inmunológico adaptativo está formado por células T y B13.
Otro mecanismo obedece a los neutrófilos, que desempeñan un papel vital en la respuesta inmune innata y son la primera línea de defensa contra enfermedades infecciosas, y su producción de especies reactivas de oxígeno. Las anomalías del estrés oxidativo mediadas por neutrófilos pueden desempeñar un papel importante en la patogénesis de la EB, y los productos proteicos de oxidación avanzada pueden ser un marcador útil para monitorizar la progresión y la gravedad de la actividad de la enfermedad en pacientes con EB. Asimismo, estos pacientes presentan niveles elevados de citocinas y quimiocinas proinflamatorias, incluidas IL-8, TNF-α, INF-γ, GM-CSF/G-CSF y CXCL-8. La EB, también conocida como vasculitis crónica, se caracteriza por trombosis venosa, aneurismas y oclusiones. A diferencia de la vasculitis clásica, los estudios patológicos en pacientes con EB han demostrado una falta de vasculitis necrosante verdadera, granuloma o depósito de inmunocomplejos12,13.
La respuesta inmune Th1 tiene un papel importante en la patogénesis de la EB. Los niveles de expresión de las células Th1 y citocinas relacionadas se asocian con la actividad de esta enfermedad. Los estudios han encontrado que las frecuencias de las células Th1 y sus citocinas y el factor de transcripción T-bet eran significativamente mayores en pacientes con EB activa que en pacientes con EB inactiva. Se han informado niveles elevados de citocinas Th1, como IL-2, IL-12, IL-18 e IFN-γ, en las células mononucleares de sangre periférica de pacientes con EB activa12,13.
IL-1, IL-6 y TNF-α son las principales citocinas proinflamatorias en pacientes con EB. Estas citocinas se han encontrado en el líquido ocular de pacientes con EB durante más de 20 años y se cree que son los principales mediadores inflamatorios que conducen al desarrollo de la enfermedad12,13.
Manifestaciones clínicasLas manifestaciones mucocutáneas son marcadores de EB y su reconocimiento puede permitir el diagnóstico y el tratamiento. Cuanto más temprana es la aparición de las manifestaciones, peor es el pronóstico, con el consiguiente aumento de la morbimortalidad2,15,16.
El cuadro clínico de EB es muy heterogéneo; se encuentran notables diferencias entre grupos etarios y entre diferentes zonas geográficas. Las manifestaciones clínicas se suelen presentar con un patrón recurrente que alterna periodos de actividad y remisión, y el curso es difícil de predecir3,7,17.
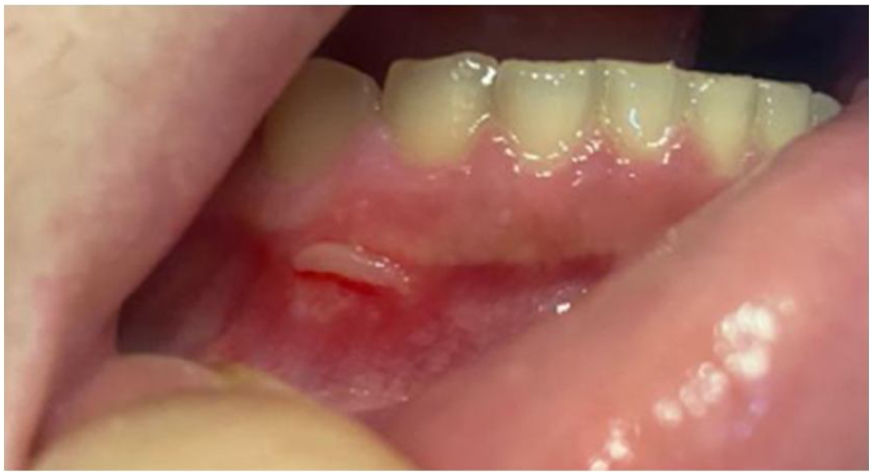
Las úlceras orales (fig. 2) están presentes en casi todos los casos. Estas lesiones son la manifestación inicial hasta en el 80% de los pacientes y preceden a las demás en una media de 7-8 años. Son úlceras dolorosas, de bordes bien definidos y halo eritematoso. También se observa la formación de una pseudomembrana amarillenta o grisácea. Se clasifican por tamaño y disposición en úlceras menores (<1cm), mayores y herpetiformes. Aparecen en brotes con un promedio de 9,8 veces al año, con úlceras menores que duran entre 7 y 10 días, y hasta 4 semanas en los casos de úlceras mayores. Se localizan preferentemente en las porciones no queratinizadas de la mucosa oral. Pueden dejar cicatrices y secuelas, como disfagia, odinofagia y disnea. Clínicamente son casi indistinguibles de la aftosis oral recurrente. Sin embargo, las características que favorecen el diagnóstico de EB incluyen el número de úlceras (>6), la aparición sincrónica de más de una variante clínica, el enantema difuso y el compromiso del paladar blando y la orofaringe. El seguimiento clínico riguroso es el mejor recurso para el esclarecimiento diagnóstico2,3,16.

Las úlceras genitales, otra manifestación clásica, son similares a las úlceras orales en apariencia y curso. Sin embargo, son menos recurrentes, tienen mayor tendencia a la formación de cicatrices y algunas presentan bordes necróticos. Las lesiones más profundas pueden complicarse con la aparición de fístulas, especialmente en las mujeres. En los hombres, el sitio más afectado es el escroto, pero las áreas afectadas pueden incluir el prepucio, el pene, el glande y, en algunos casos, el epidídimo. En las mujeres, las úlceras en la vagina y el colon pueden ser oligosintomáticas. La región más frecuentemente afectada son los labios mayores2,17,18.
Aunque las lesiones cutáneas no son específicas de la EB, son esenciales para el diagnóstico. Su frecuencia varía del 48 al 88% en los pacientes diagnosticados. Las manifestaciones cutáneas se pueden dividir en lesiones papulopustulares, lesiones de eritema nudoso (EN), tromboflebitis y lesiones cutáneas y vasculíticas variadas. Las lesiones iniciales presentan vasculitis leucocitoclástica o agresiones neutrofílicas a los vasos, mientras que las maduras se caracterizan por vasculitis linfocítica2,17,19.
Las lesiones papulopustulares son las más prevalentes y tienden a ser más frecuentes en individuos con respuesta positiva a una prueba de patergia o en aquellos con afectación articular. Suelen aparecer como pápulas que se convierten en pústulas en 24-48h. Estas lesiones son estériles y pueden parecerse a la foliculitis o tener una apariencia de acné. Este último puede ser clínica e histopatológicamente indistinguible del acné vulgar. Una pista para la diferenciación es la ausencia de comedones y la marcada afectación de las extremidades. La mala respuesta a las terapias clásicas contra el acné, como los antibióticos, es otro indicador2,3,17,19.
Las lesiones de EN ocurren en un tercio de los pacientes y generalmente afectan las extremidades inferiores. También pueden verse afectadas las regiones de los glúteos, la cara y el cuello. No evolucionan con ulceración y duran entre 2 y 3 semanas. La recurrencia es común. Se puede distinguir de las lesiones de EN clásicas por la presencia de vasculitis en los exámenes histopatológicos. Suelen presentarse con hiperpigmentación residual2,17–21.
La tromboflebitis subcutánea a menudo se confunde con lesiones de EN. Se caracteriza por nódulos eritematosos dolorosos, que en ocasiones muestran una disposición lineal. Las lesiones pueden migrar diariamente dependiendo del segmento vascular afectado. Son importantes por ser un marcador de otras afectaciones vasculares6,17,18,20.
Las úlceras cutáneas afectan hasta el 3% de los pacientes, son recurrentes y dejan cicatrices. Ocurren en varios lugares, como piernas, axilas, senos y regiones interdigitales e inguinales. Según algunos autores, son la manifestación cutánea más específica de la EB. Son comunes en los niños afectados. En este grupo de edad las úlceras perianales y las artralgias son más frecuentes y también se observa un curso clínico más grave de uveítis2,17,19,20.
Las manifestaciones vasculares pueden aparecer como púrpura palpable, infartos subungueales o síntomas que imitan cuadros clínicos de enfermedades como el síndrome de Sweet, el pioderma gangrenoso y el eritema multiforme. En estos casos, la presencia de otros signos de EB o la presencia de HLA-B51 puede ayudar al diagnóstico diferencial. Además, Schreiner y Jorizzo postularon que solo las lesiones con reacción vascular neutrofílica o vasculitis leucocitoclástica documentada pueden atribuirse a EB17,21,22.
La enfermedad ocular, más común entre los hombres, afecta a la retina y a la úvea; se presenta en un 30-70% de los pacientes y ocasiona ceguera en un 25% de ellos5. Suele aparecer 2-3 años después de las úlceras orales o genitales, pero puede ser la primera manifestación de la enfermedad en un 10-20% de los casos. Se caracteriza por episodios de uveítis crónica no granulomatosa, bilateral, recurrente, que afecta el segmento anterior, posterior o ambos ojos (panuveítis). Un tercio de los pacientes puede presentar hipopion o acumulación de pus en la cámara anterior del ojo. Otras manifestaciones incluyen iridociclitis, queratitis, epiescleritis, escleritis, vitritis, hemorragia vítrea, vasculitis retiniana, oclusión de la vena retiniana, neovascularización retiniana y neuritis óptica. Estos hallazgos están significativamente relacionados con la reducción de la agudeza visual17,21–23.
El deterioro neurológico ocurre en el 5-10% de los pacientes y afecta principalmente a hombres5. Se presenta unos 5 años después del inicio de la enfermedad y afecta principalmente al sistema nervioso central y, en menor medida, al sistema nervioso periférico. Esta afectación puede ser parenquimatosa, no parenquimatosa o ambas. En el primer caso, afecta al tronco del encéfalo o a los ganglios basales, lo que se relaciona con un peor pronóstico. La enfermedad cerebral no parenquimatosa comprende la trombosis venosa cerebral, la vasculitis arterial y la meningitis aséptica. Su pronóstico es desfavorable en todas sus formas. La resonancia magnética es importante incluso para el diagnóstico diferencial con la esclerosis múltiple6,15,21,22,24. Una serie de casos que describe 23 niños con trombosis del seno venoso cerebral causada por EB evidencia su inclusión en el diagnóstico diferencial de niños con dolor de cabeza persistente25.
Las trombosis venosas profundas de las extremidades son la forma más común de afectación vascular junto con la trombosis venosa superficial recurrente. Los hombres se ven más afectados que las mujeres. La trombosis venosa profunda de las extremidades ocurre en el 30-40% de los pacientes, y la trombosis de la vena cava inferior o superior se reporta en el 0,2-9% de los casos21,22,24.
La afectación articular se informa en el 45-60% de los pacientes con EB e incluye artralgia y monoartritis o poliartritis no erosiva y no deformante. Afecta rodillas, tobillos, caderas, codos y muñecas con infiltrados sinoviales inflamatorios neutrofílicos y mononucleares y trombosis de pequeños vasos. El tratamiento antiinflamatorio generalmente es eficaz y tiene buen pronóstico. La espondilitis anquilosante fue descrita en el 10% de los casos por Hatemi et al. La pseudofoliculitis fue la manifestación más asociada a esta afectación17,19,21.
El tracto gastrointestinal se ve afectado en un 3-26% de los pacientes, variando en diferentes poblaciones, siendo más frecuente en Japón que en Oriente Medio y el Mediterráneo. La inflamación y la ulceración de las mucosas puede ocurrir en todo el tracto gastrointestinal, pero especialmente en la región ileocecal, donde se puede observar la presencia de grandes úlceras ovaladas. Se debe hacer un diagnóstico diferencial con la enfermedad de Crohn3,21,22,24.
Pueden aparecer episodios recurrentes de fiebre en el 43% de los pacientes con EB pediátrica. Cuando ocurren, las aftas orales acompañantes no son infrecuentes. La fiebre también puede acompañar la aparición de manifestaciones vasculares, neurológicas o articulares15,18,21.
Las manifestaciones de la EB son amplias en la edad pediátrica. Al igual que en los pacientes adultos con EB, las ulceraciones orales recurrentes, que se observan en el 96-100% de los casos pediátricos, son el hallazgo más común de la enfermedad en niños. Las úlceras genitales, que se notifican en el 57-93% de los casos de adultos, suelen localizarse en el escroto o los labios mayores y menores. La afectación ocular es una de las causas más importantes de morbilidad en la EB y se informa en el 14,1-66,2% de los casos de EB pediátrica, donde encontramos uveítis anterior con hipopion, uveítis posterior, iridociclitis, queratitis, epiescleritis, hemorragia vítrea, cataratas, glaucoma y desprendimiento de retina3,17,26,27. Otras manifestaciones que se han reportado en la EB son lesiones del parénquima pulmonar (nódulos y cavidades), hepatitis, nódulos hepáticos, derrames pleurales, pericarditis, miocarditis y glomerulonefritis3,17,26.
DiagnósticoEn 1969, Mason y Barnes definieron como criterios mayores las úlceras orales, las úlceras genitales y las lesiones oculares y cutáneas, y como criterios menores, la afectación de los sistemas gastrointestinal, cardiovascular y nervioso central o la tromboflebitis, la artritis y los antecedentes familiares. Afirmaron que la presencia de 3 criterios mayores o de 2 mayores y 2 menores indicaría EB28. Ahora bien, cabe resaltar que en la actualidad los criterios utilizados en adultos fueron propuestos en 1990 y revisados en 2006 y 2014. Según estos criterios, la aparición de úlceras orales como criterio mayor y 2 de los hallazgos cutáneos y oculares establecerían el diagnóstico con una sensibilidad del 85% y una especificidad del 96%2,13,28.
Ahora bien, la sensibilidad de dichos criterios de clasificación es baja, particularmente en niños; si bien la presencia de una úlcera oral es fundamental para el diagnóstico, la falta de mención de los compromisos vasculares y neurológicos puede generar confusión en el diagnóstico. El estudio de Behçet pediátrica (PEEB) de 2015 tuvo como objetivo definir los criterios de clasificación para pacientes pediátricos utilizando la cohorte más grande (tabla 1), donde el cumplimiento de 3 criterios de los 6 en un paciente podría clasificarlo como EB pediátrica, lo que ha permitido generar una alarma que lleva a un abordaje temprano de estos pacientes3,5–7,17,26,27.
Criterios pediátricos de clasificación internacional para la enfermedad de Behçet
| Criterio | Definición | Puntaje |
|---|---|---|
| Aftosis bucal recurrente | Al menos 3 ataques/año | 1 |
| Ulceración o aftosis genital | Típicamente con cicatriz | 1 |
| Compromiso de la piel | Foliculitis necrótica, lesiones acneiformes, eritema nudoso | 1 |
| Compromiso ocular | Uveítis anterior, uveítis posterior, vasculitis retiniana | 1 |
| Signos neurológicos | Con la excepción de dolores de cabeza aislados | 1 |
| Signos vasculares | Trombosis venosa, trombosis arterial, aneurisma arterial | 1 |
Fuente: Criterios pediátricos para la enfermedad de Behçet 20156.
Se requieren 3 de 6 elementos para clasificar a un paciente con EB pediátrica. La patergia se define como un estado alterado de la respuesta del tejido a un traumatismo mínimo por punción con aguja, que refleja una respuesta exacerbada del sistema inmunológico innato. La metodología de prueba puede variar entre instituciones, pero la mayoría de los estudios aplicó una aguja de 20-26G para realizar 4-6 punciones intradérmicas en un ángulo de 45° en la región flexora del antebrazo. Este es el sitio elegido por tener una tasa de positividad más alta. La lectura debe realizarse 48h después del procedimiento. Las lesiones de patergia se manifiestan clínicamente como pápulas eritematosas, a veces rematadas por una pústula estéril. Clásicamente, la prueba se considera positiva cuando se produce una reacción superior a 2mm. Sin embargo, los exámenes histopatológicos pueden mostrar alteraciones típicas de las lesiones de pequeño diámetro. La dermatoscopia también puede ser útil para la evaluación2,12,19,21,29.
El diagnóstico de EB se basa sustancialmente en las características clínicas, pero no se describen síntomas ni signos específicos. El espectro del diagnóstico diferencial es extremadamente amplio, incluyendo enfermedades autoinmunes y autoinflamatorias3,7,26.
En niños, el diagnóstico es aún más desafiante debido al largo intervalo de tiempo entre el inicio de la enfermedad y el desarrollo de un cuadro clínico compatible con los criterios diagnósticos. Se recomienda una anamnesis detallada y un examen sistémico en la evaluación de un niño con úlceras orales o genitales (la presentación más típica de EB en niños), junto con un seguimiento crítico. Los pacientes con EB pueden no tener elevación de reactantes de fase aguda, excepto en caso de afectación cerebral o vasculitis; el hecho de tener una prueba de patergia negativa o antígeno HLA-B51− no descarta la enfermedad3,5,26,27.
En todos los pacientes con sospecha de EB se debe realizar una anamnesis completa dirigida a la detección o exclusión de sintomatología en referencia a los principales órganos o sistemas diana de la enfermedad, incluyendo analítica sanguínea: hemograma, bioquímica general, pruebas de coagulación, evaluación de reactantes de fase aguda (velocidad de sedimentación globular, proteína C reactiva), marcadores de citólisis (lactato deshidrogenasa, creatina cinasa), proteínas totales, albúmina, estudio inmunológico básico (inmunoglobulinas, C3, C4), HLA-B51, estudio básico de orina, exploración física completa por aparatos con especial atención a la exploración genital en busca de lesiones activas o cicatrices de lesiones previas, valoración oftalmológica, incluso en ausencia de síntomas oculares, y valoración cardiológica (electrocardiograma y ecocardiografía), incluso en ausencia de síntomas7,17,18,26.
En la actualidad se han descubierto múltiples imitadores monogénicos de la EB que se traducen en la pérdida de función de la línea germinal en el gen TNFAIP3, lo que lleva a una haploinsuficiencia de la proteína A20, que es una proteína reguladora en la vía de señalización NF-κB, cuya presentación clínica incluye úlceras orales y genitales, patergia, trombosis vascular y afectación neurológica y gastrointestinal, por lo que debe considerarse en pacientes con antecedentes familiares de EB, aparición temprana de síntomas y enfermedad febril recurrente7.
TratamientoLa EB es una dolencia multisistémica, con síntomas que dependen de la edad, el sexo y el origen étnico. El primer paso en el abordaje de esta heterogénea enfermedad es determinar los objetivos del tratamiento. Los objetivos principales deben ser controlar los brotes inflamatorios, que son la característica típica de esta enfermedad, así como prevenir el daño orgánico irreversible. Debe tenerse en cuenta el tipo de órgano afectado y el nivel de daño, así como la edad, el sexo y las preferencias de tratamiento del paciente (tabla 2). Dado que esta enfermedad afecta a diferentes sistemas, el tratamiento requiere un enfoque multidisciplinario5,30,31.
Terapias recomendadas para las principales manifestaciones clínicas de la enfermedad de Behçet
| Manifestaciones clínicas | Primera línea | Segunda línea | Otras opciones |
|---|---|---|---|
| Afectación mucocutánea | Corticoides tópicosColchicina (prevención) | AZAIFN-αEtanercept Infliximab Adalimumab | ApremilastaMTXAnakinra Canakinumab Ustekinumab Secukinumab Talidomida |
| Artritis | Esteroides intraarticularesColchicina | Dosis bajas de esteroidesAZAIFN-αInfliximabAdalimumab | Secukinumab |
| Compromiso ocular | CorticosteroidesbAZACiclosporina(Infliximab)d(Adalimumab)d | InfliximabAdalimumabIFN-α | Esteroides intravítreoscMTXAnakinraCanakinumabTocilizumabRituximab |
| Neuro-Behçet | CorticosteroidesAZAAnticoagulacióne(Infliximab)d(Adalimumab)d | InfliximabAdalimumab | MMFMTXTocilizumabRituximab |
| Compromiso arterial | CorticosteroidesCiclofosfamidaf | InfliximabAdalimumab | Cirugía/stent |
| Implicación del tracto gastrointestinal | Corticoides5-ASAAZA | InfliximabAdalimumab | Talidomida |
AZA: azatioprina; IFN-α: interferón alfa; MMF: micofenolato mofetilo; MTX: metotrexato; 5-ASA: 5-aminosalicilato.
Desde su introducción, los fármacos biológicos (principalmente IFN-α y anti-TNF-α) han mejorado notablemente el manejo de los pacientes con EB. El primer agente biológico introducido para la EB fue el IFN-α, por sus conocidas propiedades inmunomoduladoras. Esta citocina es efectiva para inducir la remisión en pacientes afectados por EB, con mayor eficacia en aquellos con uveítis severa. Los agentes anti-TNF-α se han utilizado en una amplia gama de manifestaciones graves de EB, que incluyen uveítis, neuro-Behçet, compromiso gastrointestinal, artritis, vasculitis y manifestaciones mucocutáneas. En particular, se ha demostrado que el infliximab y el adalimumab son efectivos en pacientes con uveítis y enfermedad gastrointestinal grave, mientras que estudios aleatorizados demostraron la eficacia de etanercept en la afectación mucocutánea16,18,31. Hoy en día existen estudios registrados en ClinicalTrials que buscan mejorar la sintomatología del paciente con EB (anexo 1).
ConclusiónLa EB es una dolencia rara pero compleja, que se puede presentar en la infancia y requiere un abordaje multidisciplinario en colaboración con pediatras, reumatólogos, dermatólogos, oftalmólogos, neurólogos, gastroenterólogos y otros especialistas cuando sea necesario. Se necesitan estudios multicéntricos, controlados con placebo y estandarizados que involucren grandes series de pacientes, utilicen puntuaciones clínicas y tengan un seguimiento a largo plazo para comprender mejor la naturaleza de esta enfermedad. Las nuevas opciones terapéuticas en desarrollo a la luz de las hipótesis patogénicas pueden ser prometedoras.
Consideraciones éticasEsta es una revisión de la literatura que se acoge a lo emanado por la Declaración de Helsinki y lo dispuesto en la Ley General de Salud en materia de investigación, por lo cual no se requiere comité de ética ni de investigación. Se tiene aval de autor intelectual de la fotografía utilizada para su publicación. Los autores declaran que este artículo no contiene información personal que permita identificar a los pacientes.
FinanciaciónNo se recibió financiación para la redacción del informe del caso.
Conflicto de interesesLos autores declaran que no tienen intereses en competencia. Este manuscrito no está siendo considerado por ninguna otra revista.
Declaración de la IA generativa y las tecnologías asistidas por IA en el proceso de escrituraLos autores declaran que no se utilizó IA ni tecnologías asistidas por IA en el proceso de investigación ni de redacción de este manuscrito.
