




La enfermedad de Camurati-Engelmann es una entidad poco común debida a mutaciones en el gen que codifica el TGF-β. Se caracteriza por hiperostosis de huesos largos y cráneo, acompañada de dolor óseo intenso, ocasionalmente debilidad muscular, marcha de pato. El tratamiento se basa en el uso de glucocorticoides en dosis altas y en casos severos la descompresión quirúrgica está indicada. Desde nuestro conocimiento este es el primer caso reportado en Colombia.
Camurati-Engelmann disease is a rare entity due to mutations in the gene encoding the TGF-β. It is characterised by hyperostosis of long bones and skull, accompanied by severe bone pain, and occasionally muscular weakness and a waddling gait. The treatment is based on the use of high doses of glucocorticoids, and in severe cases surgical decompression is indicated. As far as we know, this is the first case reported in Colombia.
La enfermedad de Camurati-Engelmann (ECE) conocida como displasia diafisiaria progresiva, es una enfermedad poco común debida a mutaciones del factor de crecimiento transformante beta (TGF-β); el cual participa en la proliferación ósea. El dolor en huesos largos es el síntoma cardinal de la ECE. No existe tratamiento curativo de la enfermedad, sin embargo, puede realizarse un ciclo de glucocorticoides en dosis altas y, ocasionalmente, pueden realizarse cirugías descompresivas del canal medular que ayudan a aliviar el dolor.
Presentamos el caso de una mujer de 54 años con ECE, con compromiso tibial sin otras manifestaciones óseas. Desde nuestro conocimiento este es el primer caso de ECE reportado en Colombia.
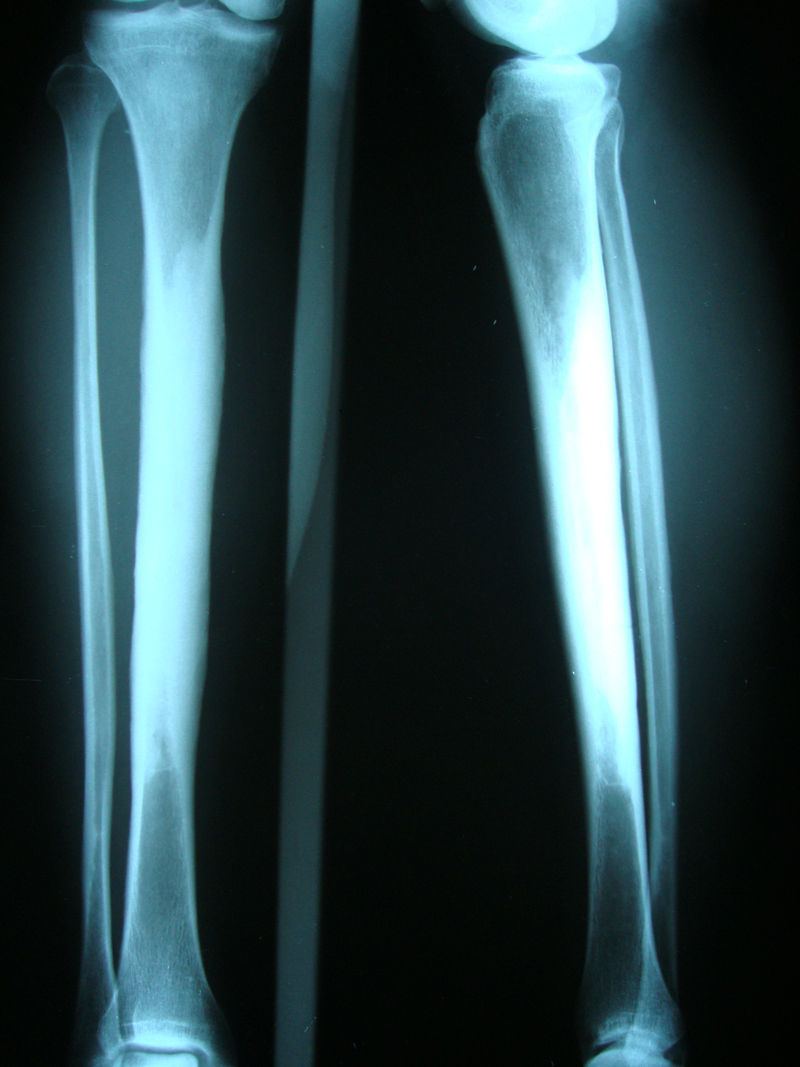
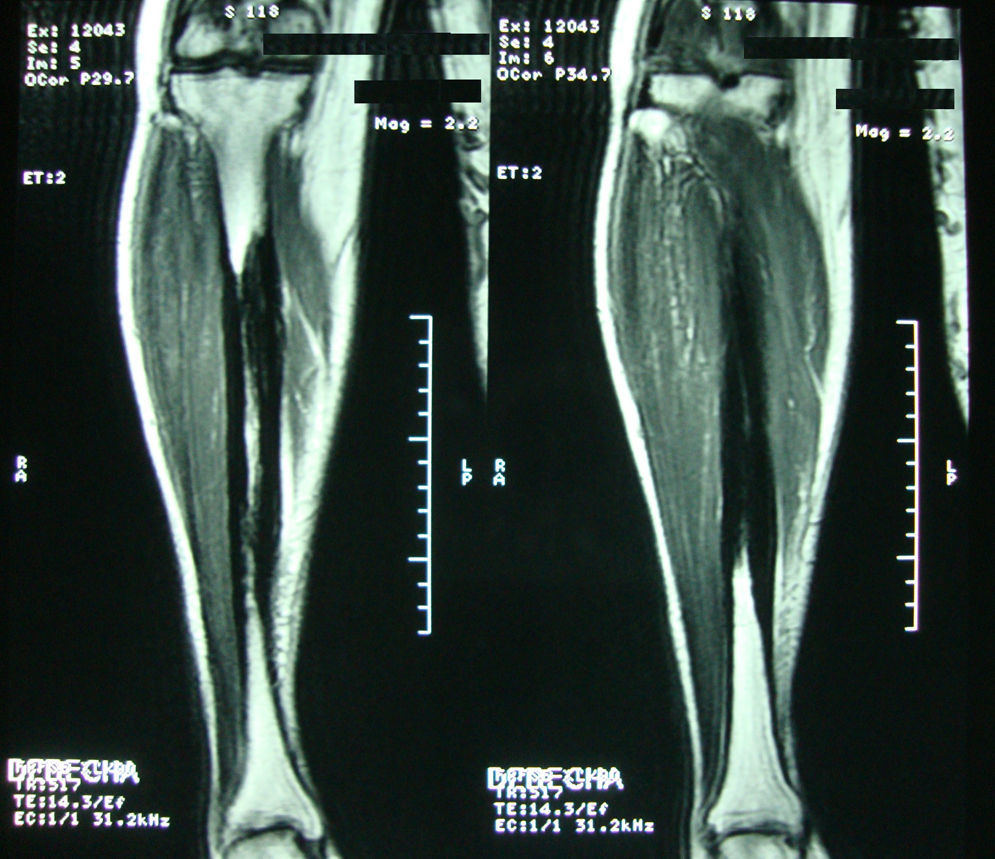
Caso clínicoMujer de 54 años, quien hace 4 años presentó trauma contundente en tibia izquierda, desde esa época presenta dolor persistente en ambos miembros inferiores. En la revisión por sistemas negaba trastornos oculares, auditivos, fracturas previas, déficit neurológico o síntomas constitucionales. Recibió tratamiento médico con AINE sin ninguna mejoría. Por lo cual se ordenó radiografía de ambas piernas que mostró lesión esclerosante diafisiaria en tercio medio con pérdida del canal medular (fig. 1). La RNM en el 2008 confirmó dichos hallazgos (fig. 2). Los datos de laboratorios como CH, VSG, PCR, BUN, creatinina, LDH, FA, P de O fueron normales. Se realizó biopsia ósea en la cual se observó la presencia de gran cantidad de pequeños fragmentos de tejido óseo denso esclerótico sin evidencia de lesión tumoral o inflamatoria. Los hallazgos anteriores son compatibles con el diagnóstico de ECE. Fue valorada por el servicio de ortopedia quienes realizaron resección de las lesiones escleróticas y recanalización de tibia izquierda.


Desde hace 2 años presenta dolor en rodillas en relación con el ejercicio, muñecas, codos, hombros, región lumbar sin signos inflamatorios, insomnio y fatiga física. Al examen físico inicial: se encuentra una paciente con talla y fenotipo normales, dolor a la percusión de huesos largos de miembros inferiores sin deformidad ósea o déficit neurológico con puntos sensibles 15/18. Se hace además un diagnóstico de fibromialgia. Los laboratorios en esta época mostraban PTH intacta, calcio, fósforo sérico, proteínas con diferencial normales con una proteína C reactiva de 96 mg/l, por lo cual se inicia tratamiento con prednisolona a 50 mg QD, calcio+vitamina D QD, sertralina 50 mg QD y trazodone 50 mg QD.
Fue valorada por endocrinología descartando hiperparatiroidismo. En los controles posteriores se realizó disminución paulatina de los glucocorticoides hasta la suspensión en 6 meses; con desaparición del dolor a la percusión de tibia y peroné, pero la paciente continuaba con signos y síntomas claros de fibromialgia. Los reactantes de fase aguda se habían normalizado. Se solicitó valoración por siquiatría para manejo de la depresión asociada. En los controles posteriores su enfermedad de base ha permanecido estacionaria.
RevisiónLa ECE ha sido conocida por los nombres de displasia epifisaria progresiva, hiperostosis generalizada, enfermedad hiperostótica múltiple congénita, displasia esclerosante y osteoesclerosis simétrica1, pero el epónimo de ECE es el nombre más ampliamente conocido y aceptado.
La primera descripción fue realizada por Cockayne en 19202, 2 años más tarde Camurati sugirió la naturaleza hereditaria3 y en 1920 Engelmann reportó un caso característico de la enfermedad4. La ECE tiene una herencia autosómica dominante con penetrancia incompleta y expresividad variable5. Recientemente, se localizó el gen responsable de la enfermedad en la región 19q13.1-13.36. Se han encontrado mutaciones en el gen que codifica el TGF- β en un 90-94%. Este gen tiene 2 exones y la mayoría de dichas mutaciones están en el exón 4, seguido del 1. Los 3 alelos mutantes más frecuentes son p Arg210cys, p Arg218His y pCys 225 Arg7.
El factor de crecimiento transformante beta (TGF- β) es un factor de crecimiento óseo miembro de la superfamilia de las proteínas morfogenéticas óseas8; promueve la proliferación, función y sobrevida de los osteoblastos9, adicionalmente, posee una acción supresora de la diferenciación de los osteoclastos10.
La ECE es considerada una enfermedad rara con una prevalencia estimada de uno en un millón de habitantes, en la literatura mundial han sido identificados 200 pacientes. Esta patología tiene distribución mundial basada en el reporte de 24 familias de América, Europa, África y Oceanía. El inicio de la enfermedad es en la niñez y habitualmente progresa durante la adolescencia, pero en la adultez su curso puede ser estacionario o lentamente progresivo. Los síntomas clínicos están presentes en el 74% de los enfermos11. La marcha claudicante es una manifestación precoz pero no la más frecuente, ocurriendo en un 43%. También puede presentarse atrofia muscular progresiva y disminución de la grasa subcutánea de las extremidades. La fatiga fácil es un síntoma que ha sido descrito en un 44%. El síntoma cardinal de la enfermedad es el dolor óseo de las extremidades en 90% de los casos. Los pacientes refieren que el dolor se incrementa con la actividad física, el estrés y el frío. Algunos enfermos presentan crisis de dolor de duración variable (horas-semanas); 58% tiene dolor a la percusión ósea. La afección craneal ocurre en el 38% de los casos y puede manifestarse por atrofia del nervio óptico, glaucoma, subluxación del globo ocular, hipoacusia neurosensorial o de conducción en el 15%12. La fisiopatología de estas lesiones son la compresión nerviosa u obstrucción del conducto auditivo externo. La esclerosis del foramen magno puede ocasionar hiperreflexia, debilidad, paraparesia e incluso la muerte13. El aumento en la densidad mineral ósea en las diáfisis puede aumentar el riesgo de fractura con retraso en la consolidación14.
Se ha descrito un fenotipo típico de la enfermedad caracterizado por craneomegalia con frente prominente, exoftalmos, extremidades delgadas con huesos gruesos y arqueados, aspecto marfanoide, pies planos y valgus, lordosis lumbar y escoliosis.
Ocasionalmente, han sido reportados hepatoesplenomegalia, fenómeno de Raynaud, hiperhidrosis de manos y pies, retraso en la dentición y en la pubertad15. En los laboratorios puede observarse anemia, leucopenia, aumento de la fosfatasa alcalina, paratohormona, calcio y fósforo sérico, velocidad de sedimentación globular, pero no es la norma encontrar dichas alteraciones16–18,14,19.
El diagnóstico de la enfermedad se basa en los síntomas clínicos y hallazgos de laboratorios, pero se confirma con las imágenes radiológicas. Las manifestaciones radiológicas características son el engrosamiento fusiforme de las corticales de los huesos. La ECE tiene afectación simétrica y compromete tanto la superficie endóstica como perióstica. Inicialmente comienza en las diáfisis y se extiende a las metáfisis, respetando las epífisis16,20,21. Como consecuencia se observa estrechamiento del canal medular en forma de frasco de Erlenmeyer. Los huesos afectados en orden de frecuencia son: fémur, tibia, peroné, húmero, cúbito y radio. Puede observarse compromiso de las mandíbulas, escápulas, clavículas, pelvis y base de cráneo. Rara vez existe compromiso de huesos del carpo, tarso y falanges. La imagen característica de los huesos largos se presenta en el 94% de los pacientes, 63% en pelvis, 54% en cráneo22.
Los glucocorticoides son la piedra angular en el tratamiento de la ECE, debido a que incrementan la apoptosis de los osteoblastos y osteocitos, también permiten la proliferación y diferenciación de los osteoclastos23,24. Contrariamente, se ha demostrado que los glucocorticoides aumentan la expresión de TGF-β, situación deletérea en estos pacientes25. Se utiliza prednisolona a 1 mg/kg/día seguido de una reducción rápida de la dosis para evitar efectos colaterales con el fin de controlar el dolor óseo y la fatiga. El losartán es un medicamento anti-TGF-β y pudiera utilizarse en pacientes intolerantes a los glucocorticoides con hipertensión arterial26. Otros medicamentos como los bifosfonatos han sido utilizados para el manejo de la ECE con resultados decepcionantes27. El tratamiento quirúrgico se reserva como mecanismo descompresivo del conducto auditivo externo. También se han reportado osteotomías y raspados del canal medular con recurrencia en algunos casos28.
En la tabla 1 se describen las principales enfermedades óseas esclerosantes. En la forma infantil maligna de la osteopetrosis los huesos son densos y frágiles. Puede haber sangrados espontáneos con anemia e infecciones recurrentes. Las complicaciones neurológicas son ceguera y sordera. Usualmente mueren en la primera década de la vida. En la forma tipo II ocurre en la adolescencia y se manifiesta por fracturas y a veces recurrentes, vértebra de sanduche y la imagen hueso dentro del hueso en el ilíaco. La compresión de nervios craneales por osteoesclerosis es rara29.
Causas principales de enfermedades óseas esclerosantes
| Osteopetrosis (Albers Schonberg) |
| Osteomesopicnosis (Maroteaux) |
| Osteopoiquilosis |
| Osteopatía estriada (enfermedad de Voorhoeve) |
| Hiperostosis endostial (Van Vuchem, síndrome de Worth, esclerosteosis) |
| Síndrome de Kenny-Caffey |
| Displasia metafisaria (enfermedad de Pyle) |
| Displasia diafisiaria progresiva (enfermedad de Camurati-Engelmann) |
| Melorheostosis |
La picnodisostosis se manifiesta en la infancia por estatura baja y cráneo desproporcionadamente grande. Las manos son cuadradas con dedos pequeños y tórax excavatum. Las fracturas recurrentes afectan, principalmente, los miembros inferiores30.
Osteopoiquilosis significa huesos moteados. Se confunden con metástasis óseas de cáncer de próstata y seno31. Se encuentra de manera incidental por la presencia de lesiones circulares de osteoesclerosis. Afecta las partes terminales de los huesos cortos, metáfisis y epífisis de los huesos largos32. A veces se asocia a dermatofibrosis lenticular diseminada. La etiología de la enfermedad es por mutaciones del gen LEMD333.
En la enfermedad de Van Buchem hay crecimiento asimétrico de la mandíbula durante la pubertad. No hay fracturas pero sí puede haber dolor a la percusión de huesos largos. Las alteraciones neurológicas por compresión de nervios craneales son frecuentes. La enfermedad progresa con la edad, por lo tanto es más frecuente encontrar hallazgos radiológicos en pacientes mayores. En la forma de esclerosteosis (enfermedad de Truswell-Hansen) los pacientes son altos y gordos con sindactilia y displasia ungueal. La hiperostosis clásicamente es endostial. En el síndrome de Worth ocurren los mismos hallazgos pero se diferencia de las otras 2 variantes por la presencia de mandíbula grande y puntiaguda con frente ensanchada34.
La presencia de estrías lineales en los huesos largos es característica de la osteopatía estriata; puede observarse esclerosis craneal con parálisis de nervios35.
En la melorheostosis se observa una hiperostosis excéntrica e irregular de la corteza y del canal medular, como la cera que escurre por el lado de una vela36.
En la enfermedad de Caffey-Kenny hay osificación intramembranosa. El hallazgo radiológico principal es el engrosamiento cortical interno con falta de diferenciación de las tablas craneales. Los hallazgos clínicos principales son: estatura corta, palidez de mucosas y microftalmía37.
La enfermedad de Pyle se caracteriza por ensanchamiento de las metáfisis de los huesos largos llamada deformidad de Erlenmeyer, con crecimiento de las partes mediales de las clavículas, isquion y pubis38.
Finalmente, el dolor lumbar que comienza en la adolescencia es el hallazgo clínico de la osteomesopicnosis, por esclerosis de los platillos terminales. Una lesión quística en la diáfisis del fémur ha sido descrita. Otros huesos largos o craneales son de características normales39.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
