





La enfermedad de Pompe o glucogenosis tipo ii es un trastorno autosómico recesivo, debido a la deficiencia de la enzima lisosomal α-glucosidasa ácida encargada de degradar glucógeno a glucosa. La forma de inicio en el adulto es rara y se caracteriza fundamentalmente por acumulación de glucógeno en tejido muscular estriado, cardiaco y liso. Causa debilidad muscular de predominio proximal, por lo que se puede confundir con una miopatía inflamatoria. Se presenta el caso de un adulto de 60 años con diagnóstico previo de polimiositis, en quien se confirmó una enfermedad de Pompe con demostración del déficit enzimático en sustrato biológico y se logró realizar una identificación genética.
Pompe disease, or glycogen storage disease type ii, is an autosomal recessive disorder due to the deficiency of lysosomal acid α-glucosidase, the enzyme responsible for degrading glycogen to glucose. The onset form in the adult is rare and is characterised, primarily by accumulation of glycogen in striated, cardiac, and smooth muscle tissue. It causes muscle weakness of proximal predominance, so it can be confused with an inflammatory myopathy. The case is presented of a 60 year-old adult with a previous diagnosis of polymyositis in whom Pompe disease was confirmed with a demonstration of the enzymatic deficit in a biological substrate and a genetic identification was obtained.
La enfermedad de Pompe (glucogenosis tipo ii, OMIM 232300) es una enfermedad rara, de herencia autosómica recesiva, descrita inicialmente en 1932 por el patólogo holandés Johannes C. Pompe1,2. Es causada por la deficiencia genética de la enzima lisosomal α-glucosidasa ácida (AGA), que lleva a la acumulación anormal de glucógeno en las células, con disfunción de múltiples órganos pero principalmente el músculo3,4. La acumulación muscular de glucógeno en el músculo estriado produce que los pacientes tengan disfunción muscular manifiesta típicamente por debilidad muscular de predominio proximal, siendo un diagnóstico diferencial de otras miopatías. El compromiso muscular se puede extender a los músculos respiratorios generando parálisis diafragmática, hipoventilación alveolar y, en algunos casos, falla respiratoria y muerte3,5,6. También se ha documentado acumulación cardiaca con manifestaciones como hipertrofia cardiaca y trastornos del ritmo cardiaco3,7,8. La enfermedad tiene una presentación infantil pero también existe presentación tardía (edad mayor de un año), que es un reto diagnóstico4,7,8. Los pacientes con la forma clásica infantil manifiestan la enfermedad en los primeros meses de vida con miocardiopatía hipertrófica, hipotonía significativa y debilidad muscular esquelética y, si no son tratados, mueren típicamente de insuficiencia cardiaca durante el primer año de vida6,8. Los pacientes con la forma de aparición tardía presentan un fenotipo más leve, que típicamente consiste en debilidad muscular esquelética proximal con miopatía progresiva lenta, pero rara vez tienen afectación cardiaca4,6,7,9. La aparición temprana de la enfermedad resulta de una deficiencia completa o casi completa de la proteína AGA funcional, mientras que los pacientes de inicio tardío mantienen algo de actividad enzimática residual2. La forma tardía y en especial cuando se presenta después de la quinta década es poco común6.
Caso clínicoPaciente masculino de 60 años, quien tenía un diagnóstico establecido de una miopatía autoinmune de tipo polimiositis, tenía 3 de 4 criterios de Bohan y Peter, sustentado en debilidad muscular progresiva, alteraciones electromiográficas con patrón miopático y elevación de enzimas musculares. Recibía manejo con prednisolona 30mg cada día, azatioprina 75mg cada día y fisioterapia. El paciente manifestaba pobre respuesta al tratamiento instaurado, por lo que decidió consultar.
El examen clínico mostró importante atrofia en los músculos anterior y posterior del brazo, en muslos y caderas. La fuerza muscular en miembros superiores estaba disminuida: proximal 3/5 y 4/5 distal. En miembros inferiores también se observó debilidad 2/5 proximal y 2/5 distal, además de hiporreflexia patelar. No había déficit sensitivo. El signo de Gowers fue positivo. El resto del examen clínico y los parámetros vitales fueron normales.
Por la pobre respuesta que presentaba el paciente al tratamiento recibido se decidió replantear el diagnóstico y realizar nuevos estudios (tabla 1), que incluyeron un perfil metabólico que fue normal, una electromiografía de 4 extremidades y una biopsia muscular. La electromiografía fue anormal, mostrando alteraciones compatibles con distrofia muscular progresiva y se observaron abundantes signos de hiperexcitabilidad de la membrana celular, característicos de miopatías inflamatorias. La biopsia muscular del tercio distal del bíceps del brazo izquierdo mostró pérdida de fibras musculares con reemplazo por tejido adiposo. No se observó inflamación al realizar estudios de inmunohistoquímica. Tampoco se encontraron infiltrados que indicaran una miopatía inflamatoria. Tinciones de periodic acid schift (PAS) no se realizaron.
Resultados de exámenes de laboratorio generales
| Hemoleucograma | Leucocitos: 8.350/mm3, neutrófilos: 5.835/mm3 (70%), linfocitos: 2.087/mm3 (25%), eosinófilos: 250/mm3 (3%), hemoglobina: 10,8 g/dl, VCM: 86 fL, HCM: 29 pg, plaquetas: 256.000/mm3 |
| Estudios hormonales | TSH: 1,21 mIU/l T4 libre: 0,9 ng/dl, PTH: 45 pg/ml |
| Función renal | Creatinina: 0,9 mg/dl, nitrógeno ureico: 15 mg/dl |
| Electrolitos | Sodio: 142 mEq/l, potasio: 3,9 mEq/l, cloro: 97 mmol/l |
| Enzimas musculares | CK: 69 U/l, aldolasa: 2,5 U/l, LDH: 126 U/l |
| Aminotransferasas | AST: 26 U/l, ALT 39 U/l |
| Perfil inmunitario | ANAS: negativo, ENAS: negativo |
ALT: alanina transaminasa; ANAS: anticuerpos antinucleares; AST: aspartato aminotransferasa; CK: creatina fosfocinasa; ENAS: anticuerpos antinucleares extraíbles del núcleo; LDH: deshidrogenasa láctica; PTH: hormona paratiroidea; TSH: hormona estimulante de la tiroides.
Ante la incertidumbre diagnóstica, se realizó una tamización para enfermedad de Pompe con gota de sangre en papel de filtro, la cual estaba alterada, observándose una relación α glucosidasa neutra/inhibida alta y porcentaje de inhibición de la AGA alto. Teniendo en cuenta el examen de tamización positivo, se realizó un estudio enzimático en leucocitos con relación neutra/inhibida que también estuvo alterada, lo cual confirmó una alteración de la AGA (tabla 2).
Estudios realizados en paciente para confirmar enfermedad de Pompe
| Exámenes enzimáticos para enfermedad de Pompe | |
|---|---|
| Sangre en papel filtro | Relación AGA neutra/inhibida: 20,8 (valor de referencia: menor de 16) |
| Porcentaje de inhibición de la AGA (%): 89,1 (valor de referencia: menor del 86%) | |
| Estudio enzimático en leucocitos | Relación AGA neutra/inhibida: 18,3 (valor de referencia: menor a 15) |
| Porcentaje de inhibición: 85,0 (valor de referencia: menor del 85%) | |
| Estudio en sustrato anatómico (glucógeno) | Nivel de 0,04 mmol/mg prot/h control: en 0,4-1,4 |
| Estudio de ADN y detección de mutación para enfermedad de Pompe | |
|---|---|
| Localización citogenética | 17q25.2-q25.3 |
| Exón/intrón | Intrón 1 |
| Cambio de nucleótido | c.-32-12T>G |
| Cigosidad | Homocigota |
| Base de datos y número | HGMD número: CS941489 |
AGA: α-glucosidasa ácida; HGMD: Human Gene Mutation Database.
Finalmente, se decidió realizar un estudio en sustrato anatómico (glucógeno), que mostró un nivel anormalmente bajo, con lo cual se confirmó el diagnóstico de enfermedad de Pompe del adulto. Adicionalmente, se realizó estudio genético que mostró alteración c.-32-12T>G en el estado homocigótico del gen de la AGA intrón 1 (tabla 2). Tras el diagnóstico, se realizó manejo de sustitución enzimática con α-glucosidasa recombinante humana, con dosis de 20mg/kg por perfusión por vía intravenosa lenta cada 15 días. Tras el inicio de la terapia enzimática sustitutiva, no solo se observó estabilidad de la enfermedad, sino también mejoría de la debilidad a los 3 meses de seguimiento, mostrando fuerza de 3/5 de manera bilateral en los miembros inferiores. El paciente continúa en seguimiento clínico por reumatología.
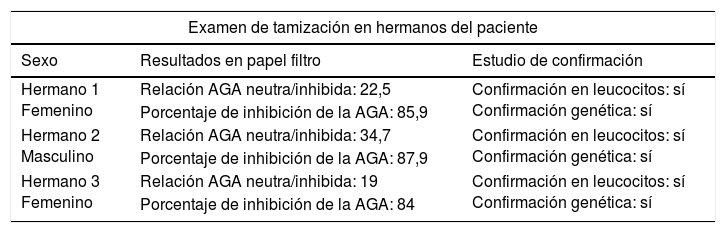
Teniendo en cuenta el carácter genético de la enfermedad, se realizó tamización en 10 hermanos del paciente, lográndose detectar alteración en la función enzimática en 3 de ellos (tabla 3). También se realizaron un estudio confirmatorio en prueba de leucocitos y un estudio genético que confirmaron la alteración y la mutación de la AGA; sin embargo, en el momento de la realización del examen ninguno manifestó síntomas.
Resultados de exámenes de tamización en hermanos del paciente
| Examen de tamización en hermanos del paciente | ||
|---|---|---|
| Sexo | Resultados en papel filtro | Estudio de confirmación |
| Hermano 1 Femenino | Relación AGA neutra/inhibida: 22,5 | Confirmación en leucocitos: sí Confirmación genética: sí |
| Porcentaje de inhibición de la AGA: 85,9 | ||
| Hermano 2 Masculino | Relación AGA neutra/inhibida: 34,7 | Confirmación en leucocitos: sí Confirmación genética: sí |
| Porcentaje de inhibición de la AGA: 87,9 | ||
| Hermano 3 Femenino | Relación AGA neutra/inhibida: 19 | Confirmación en leucocitos: sí Confirmación genética: sí |
| Porcentaje de inhibición de la AGA: 84 | ||
Relación AGA neutra/inhibida con valor de referencia menor de 16.
Porcentaje de inhibición de la AGA con valor de referencia menor del 86%.
La enfermedad de Pompe es considerada una enfermedad rara, con una incidencia entre 1:14.000 y 1:300.000, dependiendo del área geográfica estudiada, siendo la forma infantil mayor entre chinos, mientras que la forma adulta de aparición tardía tiene una incidencia más alta en Holanda7,10. El diagnóstico es difícil y suele tener un retraso entre los 4-30 años después del inicio de los síntomas3. La edad de inicio puede oscilar entre < 1 a 52 años y la edad de diagnóstico puede variar de < 1 a 78 años9. En el caso presentado, el paciente estaba diagnosticado con una polimiositis pero no tenía buena respuesta al manejo instaurado, por lo cual se buscaron alternativas diagnósticas, las cuales incluyeron: miopatías congénitas, inflamatorias (diferentes a polimiositis, cuerpos de inclusión, dermatomiositis), endocrinas, distrofias musculares (principalmente la distrofia de Duchene, la distrofia de Becker y la distrofia de cinturas), mitocondriales y miopatías metabólicas (enfermedad de McArdle, deficiencia de carnitin palmitoil-transferasa tipo ii, enfermedad de Pompe)8,11. Otros diagnósticos diferenciales que se pueden incluir son las enfermedades de motoneurona superior e inferior, que pueden cursar también con atrofia y debilidad muscular12. Tras una prueba de tamización positiva en papel filtro con gota de sangre, se pudo enfocar hacia un déficit de AGA.
Las manifestaciones clínicas de la enfermedad de Pompe del adulto son variadas, siendo la más frecuente la debilidad muscular proximal, debilidad muscular del tronco, intolerancia al ejercicio, dificultad para respirar por debilidad de músculos respiratorios (hasta en un 80%), tos inefectiva, dificultad para la marcha y dificultad para ponerse de pie tras estar en cuclillas (conocido como signo de Gowers). Sin embargo, puede tener manifestaciones en múltiples órganos que incluyen el corazón (cardiopatía hipertrófica, taquicardia supraventricular y síndrome de Wolff-Parkinson-White), el sistema nervioso (neuropatía de fibra pequeña, pérdida de audición), el sistema vascular (aneurismas cerebrales, dolicoectasia basilar), el sistema óseo (osteopenia, osteoporosis, escoliosis) y el gastrointestinal (diarrea crónica, incontinencia fecal,)2,3,6,7,11. Desde el punto de vista bioquímico, un porcentaje importante de pacientes se puede presentar con elevación de la creatina-fosfocinasa (CK) y de otras enzimas musculares, lo cual enmascara el diagnóstico y los pacientes pueden ser diagnosticados erróneamente de miopatías inflamatorias por varios años13.
El estándar diagnóstico para la enfermedad de Pompe es la demostración de la deficiencia de la actividad enzimática de la AGA4,7,8,14. La medición bioquímica se puede realizar en muestra de fibroblastos de piel cultivados, tejido de biopsia muscular o muestras de sangre sobre papel de filtro, linfocitos purificados y leucocitos mixtos4,7,10,15. Otras formas de diagnóstico que se pueden establecer es a través de una biopsia muscular con tinción de PAS positiva, teniendo en cuenta que el contenido de glucógeno muscular se eleva hasta 10 veces por encima de lo normal en la enfermedad de Pompe infantil y en menor medida en pacientes de inicio tardío4,6,7.
El gen que codifica la AGA se encuentra localizado en el cromosoma 17q25.3 y su mutación se asocia a la enfermedad autosómica recesiva de Pompe2,6,8,9. Más de 350 mutaciones de la enfermedad se han descrito2,9,16. Entre las mutaciones recurrentes en los casos de inicio infantil se encuentra una deleción Δ525T, que se observa en el 9% de los casos en Estados Unidos y en 34% de los casos holandeses, y la mutación de sitio de empalme IVS1 (-13T->G) representa aproximadamente el 50% de los casos de inicio tardío en pacientes caucásicos7,8. En nuestro caso, se documentó la mutación intrónica c.-32-12T>G en el estado homocigótico del gen de la AGA, que corresponde con la mutación patogénica más frecuente para enfermedad de Pompe de inicio tardío. Aunque esta mutación se documentó por primera vez hace muchos años (en 1994 por Huie et al.), el mecanismo por el que afecta a la codificación de la AGA no está claro hasta la fecha17.
Una vez se diagnostica un paciente con enfermedad de Pompe, es ideal tener un estudio básico de la estructura y la función cardiopulmonar, indicándose en las guías de manejo la realización de una radiografía de tórax, un electrocardiograma y un ecocardiograma en la primera visita. En las formas del adulto se recomienda idealmente la realización de pruebas de función pulmonar con espirometría, volúmenes pulmonares con medición de la presión espiratoria máxima y la presión inspiratoria máxima. También es importante brindar a los pacientes un plan de rehabilitación integral que permita reservar la función motora y fisiológica, prevenir o minimizar las complicaciones secundarias y maximizar los beneficios de la terapia de reemplazo enzimático (TRE)6,7,11.
La TRE fue aprobada en 2007 para pacientes con enfermedad de Pompe de inicio temprano y en 2010 para la presentación tardía8,11. Dicha terapia ha mostrado beneficios, en especial cuando se inicia tempranamente2,4,10,18. La terapia con la enzima recombinante AGA humana (Myozyme®) tiene una dosis estándar de 15-20mg/kg cada 15 días en infusión continua por 4 h2,11.
Dado que se trata de una enfermedad de carácter autosómico recesivo, la probabilidad de transmisión de la enfermedad de un padre a un hijo es de aproximadamente el 25%, por lo tanto, se debe realizar un asesoramiento genético7,8,11. También se justifica la búsqueda activa de la enfermedad entre hermanos, como en este caso, donde se logró determinar la alteración enzimática en 3 de 10 hermanos; sin embargo, pueden estar asintomáticos o ligeramente sintomáticos debido a la variabilidad clínica intrafamiliar8,11. Actualmente, la evidencia es insuficiente para tratar la enfermedad presintomática o las conductas que se deben realizar en los portadores de la mutación14.
ConclusiónLa enfermedad de Pompe de inicio tardío es una enfermedad rara, de baja sospecha, que puede pasar desapercibida si no se tiene en cuenta dentro de los diagnósticos diferenciales de las miopatías inflamatorias, ya que puede presentar manifestaciones clínicas similares con debilidad muscular proximal y elevación de enzimas musculares. Su detección temprana es de importancia para iniciar una TRE que permita detener el deterioro clínico y evitar las complicaciones derivadas mejorando la calidad de vida de los pacientes.
Conflicto de interesesNo hay conflicto de intereses.
