




La esclerodermia localizada juvenil es una enfermedad polimórfica que ocurre con mayor frecuencia en niñas. Se acompaña de morbilidad importante. El compromiso extradérmico es frecuente y se reportan tasas de poliautoinmunidad de hasta 7%. Al momento, se desconocen las características clínicas de los pacientes colombianos con esta enfermedad.
ObjetivoDescribir las características clínicas, morbilidades y secuelas en pacientes con diagnóstico de esclerodermia localizada juvenil, en múltiples centros de reumatología pediátrica en Colombia.
Materiales y métodosEstudio descriptivo, retrospectivo y multicéntrico. Pacientes con diagnóstico de esclerodermia localizada juvenil con un mínimo de 1 año de evolución y 6 meses de seguimiento, en 10 centros de reumatología pediátrica mediante revisión de historias clínicas.
ResultadosEl n=88. La distribución por género fue: femenino 2,1; masculino 1. Edad promedio al inicio de la enfermedad 7,1 años (0-14). Promedio de duración de la enfermedad al diagnóstico 16,5 meses (1-96). La distribución por subtipos fue morfea circunscrita (32,9%), mixta (31,8%), linear (21,5%, asciende a 55% al incluir formas mixtas con lesiones lineares) generalizada (11,4%) y panesclerótica (2,3%). Se detectaron alteraciones estéticas en el 91%, alteraciones del crecimiento en 41% y compromiso funcional de articulaciones vecinas en 32%. Se presentó compromiso extradérmico en 22,7% y poliautoinmunidad en 12,5%.
ConclusionesLa esclerodermia localizada juvenil es una enfermedad polimórfica e impredecible. En la mayoría de los casos el diagnóstico es tardío. La tasa de compromiso extradérmico sugiere que no es una enfermedad limitada a la piel. Un diagnóstico temprano, tratamiento dinámico y seguimiento cercano permiten prevenir y detectar tempranamente complicaciones derivadas de la enfermedad.
Juvenile localized scleroderma is a polymorphic disease. It is more prevalent in girls and has a significant morbidity. Extra-cutaneous involvement is common, and polyautoimmunity can reach 7%. The clinical characteristics of this disease in Colombian patients are currently unknown.
ObjectiveTo describe the clinical characteristics, morbidity and outcomes in patients with juvenile localized scleroderma in different paediatric rheumatology clinics in Colombia.
Materials and methodsA descriptive, retrospective, and multicentre study was conducted on patients with juvenile localized scleroderma with a minimum of 1 year of disease onset, and 6 months of follow-up in 10 paediatric rheumatology clinics.
ResultsThe study included 88 patients, with a gender distribution of female 2.1: male 1. Mean age at disease onset was 7.1 years (0-14). Mean disease duration at diagnosis was 16.5 months (1-96). Sub-type distribution was, circumscribed (32.9%), mixed (31.8%), and linear (21.5%, that increased to 55% if linear lesions of the mixed subtype are included), generalised (11.4%), and pan-sclerotic morphea (2.3%). Aesthetic compromise was detected in 91%, with growth disturbances in 41%, and joint functional compromise in 32%. Extra-cutaneous involvement occurred in 22.7% and polyautoimmunity in 12.5%.
ConclusionsJuvenile localized scleroderma is a polymorphic and unpredictable disease. It diagnosed late in most of the cases. Extra-cutaneous involvement suggests that is not a disease limited to skin. An early diagnosis, a dynamic treatment and a close follow-up helps to prevent, and detect, complications arising from the disease.
La esclerodermia es una enfermedad autoinmune, polimórfica, caracterizada por la presencia de esclerosis cutánea secundaria a la acumulación excesiva de colágeno1,2. Se clasifica en 2 grandes grupos, la esclerodermia localizada y la esclerodermia sistémica. Se denomina esclerodermia localizada cuando el compromiso en piel no se acompaña de afectación de órganos internos. En algunos casos puede comprometer estructuras vecinas o incluso originar síntomas a distancia, pero estos a diferencia de la forma sistémica, usualmente no comprometen órganos vitales y generan una morbilidad diferente. La esclerodermia sistémica se caracteriza por el compromiso de órganos internos y un peor pronóstico1.
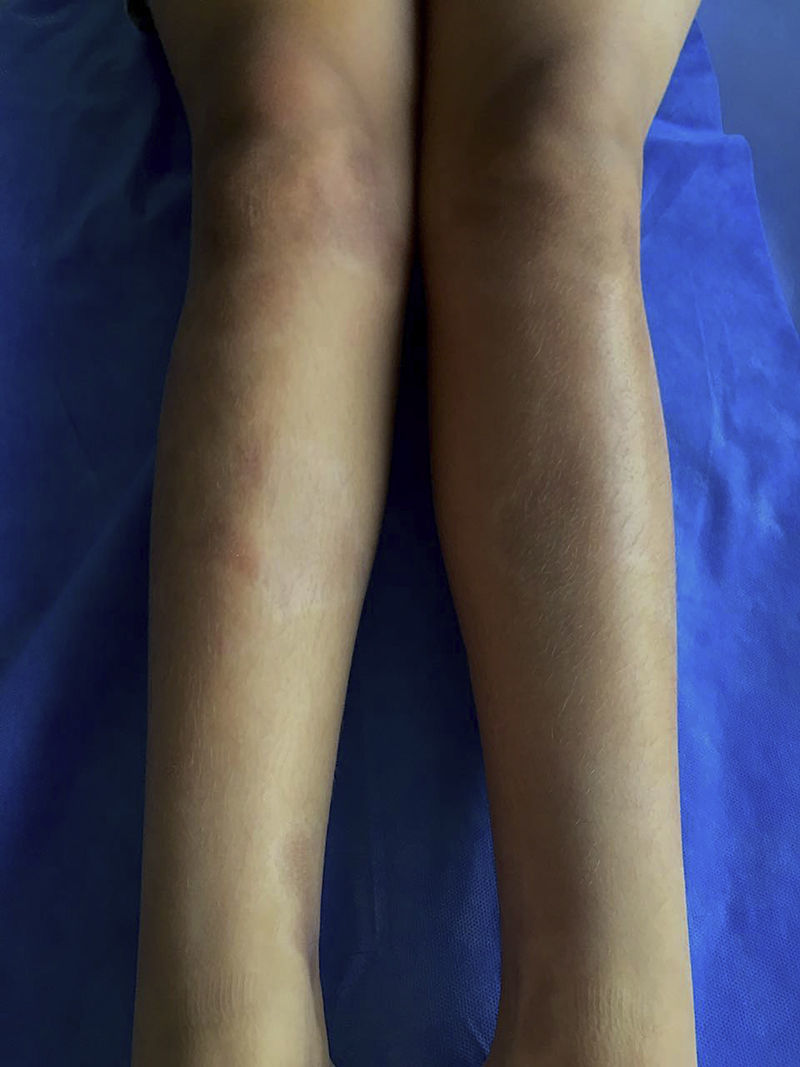
Los criterios de clasificación publicados por la Sociedad Europea de Reumatología Pediátrica (PRes) diferencian a la esclerodermia localizada juvenil (jLS) en 5 tipos según el compromiso en piel, estos son: esclerodermia linear, circunscrita, mixta, generalizada y panesclerótica. A su vez, la forma circunscrita se subdivide en las formas superficial y profunda. La esclerodermia linear puede comprometer tronco o extremidades (fig. 1) o ubicarse en cabeza donde se denomina Coup de Sabre (CDS)3. La esclerodermia localizada afecta con mayor frecuencia al género femenino4-8 y su espectro de presentación y curso clínico son muy variados.

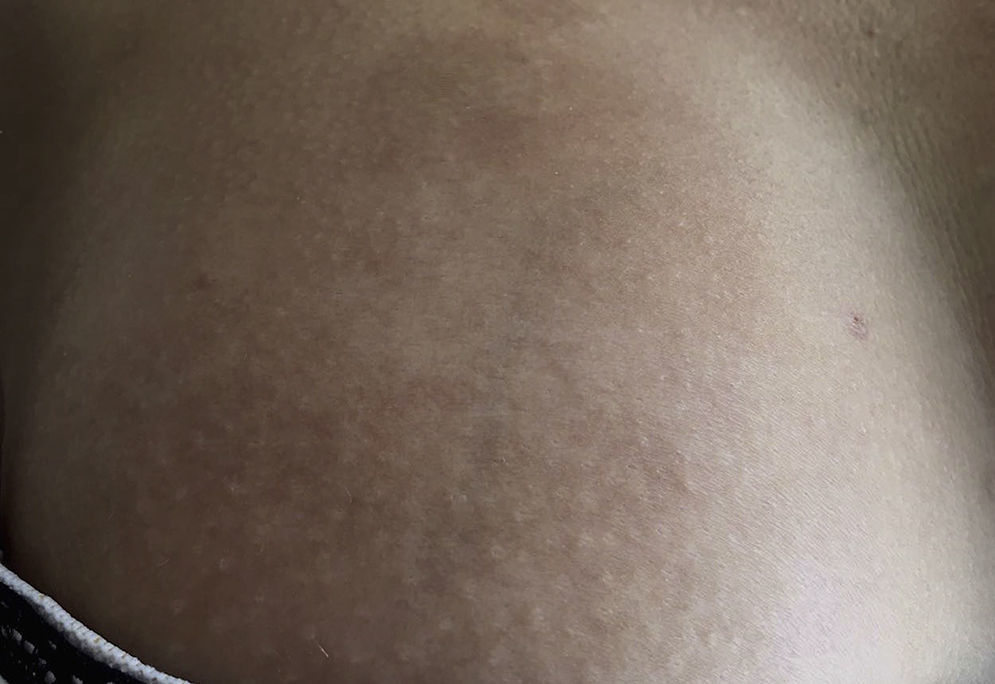
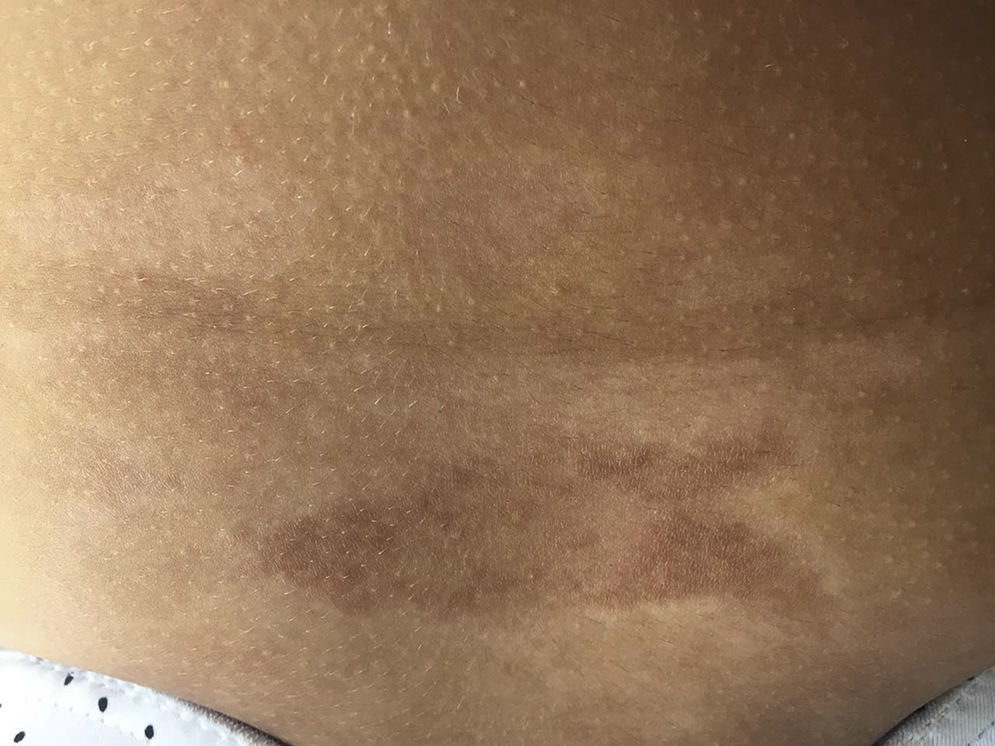
Las lesiones tempranas (fase inflamatoria) se caracterizan por un color eritematoso, violáceo y brillante con un grosor de la piel que al inicio es normal (fig. 2). Con el tiempo la fibrosis se torna más prominente con evidencia de piel indurada, hiperpigmentada y en algunos casos atrófica9 (fig. 3).


La morfea circunscrita puede mostrar mejoría con el uso de tratamiento tópico, sin embargo, debe realizarse un seguimiento cercano dado el riesgo de progresión de las lesiones y la necesidad de inicio de terapia sistémica. Dentro de las opciones terapéuticas se encuentran los corticoides tópicos, el tacrolimus y el imiquimod9. Se debe evitar el uso de esteroides tópicos en cara por tiempos prolongados. Si al completar un máximo de 3 meses de tratamiento tópico no hay mejoría, debe iniciarse terapia sistémica.
Las otras formas de esclerodermia localizada requieren un manejo combinado con esteroides sistémicos y fármacos modificadores de la enfermedad (FARMES), por un mínimo de 24 meses para disminuir el riesgo de recaídas. El FARME más utilizado es el metotrexate y ante refractariedad o intolerancia a este podría utilizarse el micofenolato mofetilo en monoterapia o en terapia combinada con metotrexate. Otros medicamentos como la inmunoglobulina intravenosa, infliximab, rituximab, ciclosporina y dapsona han sido utilizados en casos refractarios con respuestas variables. La fototerapia ha sido utilizada como terapia coadyuvante por su efecto antifibrótico e inmunosupresor. Deben tenerse siempre en cuenta los efectos como envejecimiento prematuro, carcinogénesis y la falta de efectividad en lesiones profundas1,2,9–12.
El curso de la esclerodermia localizada es impredecible, es por esto que la identificación temprana de los pacientes y el inicio de un tratamiento oportuno, integral y dinámico, está orientado a frenar la progresión de las lesiones y evitar las complicaciones. Hasta la fecha no se conocen las características clínicas de los pacientes colombianos con jLS. Conocer el comportamiento de la enfermedad en pacientes colombianos permitirá un abordaje más específico y oportuno en los pacientes.
El objetivo de este estudio es describir las características clínicas, morbilidades asociadas y secuelas en un grupo de pacientes con diagnóstico definitivo de jLS seguidos en centros de reumatología pediátrica de las ciudades de Bogotá, Cali, Cartagena y Barranquilla en Colombia.
MétodosSe realizó un estudio descriptivo, retrospectivo y multicéntrico. En una población de pacientes con diagnóstico de jLS seguidos en 10 centros de reumatología pediátrica en Colombia, mediante revisión de historias clínicas. Los criterios de selección fueron todos los pacientes que cumplieran mínimo un año de evolución de la enfermedad y mínimo 6 meses de seguimiento.
Dada la baja prevalencia de la enfermedad se incluyó el universo de los pacientes que cumplieron los criterios de selección.
Las variables relacionadas con morbilidad fueron el compromiso funcional de articulaciones vecinas, discromía, alopecia y lesiones desfigurantes, definida cuando además de los cambios discrómicos se originaron alteraciones en profundidad asociadas o no a alteraciones del crecimiento que fueron persistentes pese al tratamiento. Se definió secuela como las complicaciones permanentes relacionadas con las lesiones. Aquí se incluyen las alteraciones del crecimiento, las cuales pueden ser de tipo circunferencial, longitudinal o mixto.
Se evaluó la presencia de compromiso extradérmico y de otras enfermedades autoinmunes asociadas.
Los datos se digitaron en Excel versión 15.13.3 y el análisis estadístico se realizó en el programa SPSS versión 15.0. Las variables cualitativas se analizaron por medio de frecuencias absolutas y porcentajes, y las variables cuantitativas se analizaron por medio de promedios, desviaciones estándar, mínimos y máximos. La significación estadística se obtuvo mediante la prueba chi-cuadrado.
ResultadosSe incluyeron un total de 88 pacientes seguidos en 10 servicios ambulatorios de consulta externa de reumatología pediátrica que cumplieron los criterios de inclusión.
Características demográficasEl género femenino correspondió al 68% de los casos. La edad promedio al inicio de la enfermedad fue de 7,1 años (0 a 14 años).
El tiempo de seguimiento promedio fue de 43 meses (6 a 243 meses). El promedio de la duración de la enfermedad al diagnóstico fue de 16,5 meses (1 a 96 meses). El tiempo promedio entre el diagnóstico y la remisión a reumatología pediátrica fue de 17,1 meses (0 a 156 meses). El 98% presentó biopsia compatible con esclerodermia. Los 2 pacientes restantes presentaron lesiones en cara muy sugestivas de esta enfermedad y la biopsia fue omitida.
Según la clasificación PRes la distribución por frecuencia de mayor a menor fue: morfea circunscrita, formas mixtas, esclerodermia linear, generalizada y panesclerótica. Si se incluyen las formas mixtas que incluyen lesiones lineares, la esclerodermia linear fue la más frecuente. La mayoría de los pacientes presentaron lesiones múltiples (n=59, 67%).
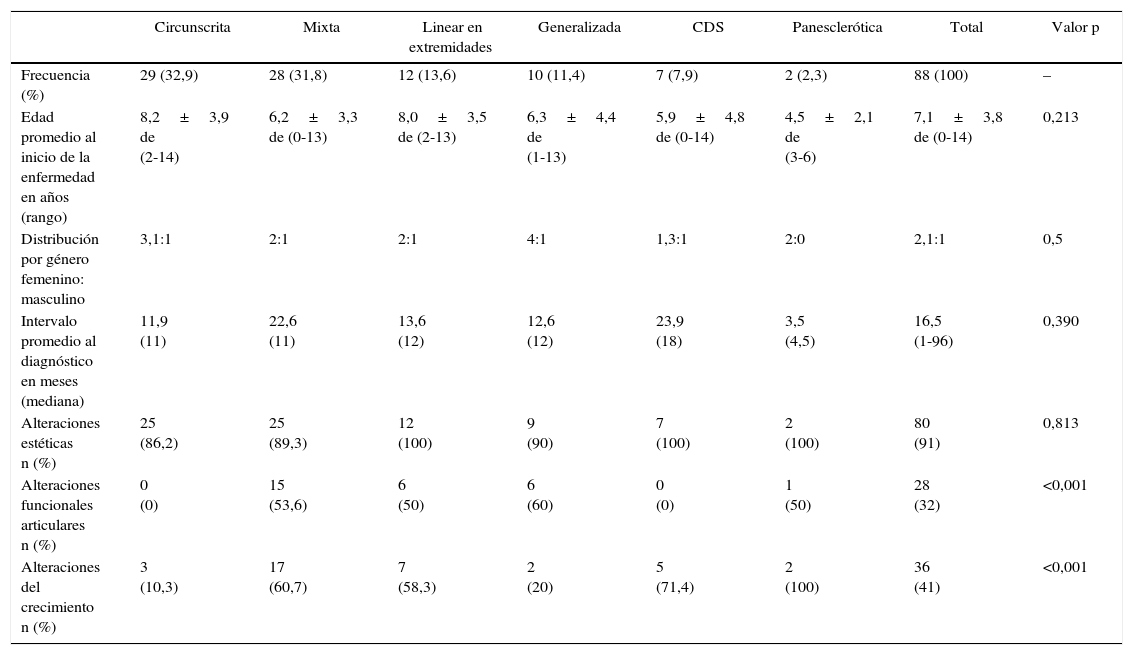
Durante el seguimiento se detectaron las siguientes morbilidades: alteraciones estéticas en el 91% de los pacientes, alteraciones del crecimiento en 41% y compromiso funcional de articulaciones vecinas en 32%. Las formas circunscrita y CDS no se asociaron a alteraciones funcionales de articulaciones vecinas. Los subtipos mixta, linear, CDS y panesclerótica, cursaron con las mayores tasas de alteraciones del crecimiento, siendo esto estadísticamente significativo. Las características según clasificación PRes se resumen en la tabla 1.
Fenotipos de esclerodermia localizada según clasificación PRes
| Circunscrita | Mixta | Linear en extremidades | Generalizada | CDS | Panesclerótica | Total | Valor p | |
|---|---|---|---|---|---|---|---|---|
| Frecuencia (%) | 29 (32,9) | 28 (31,8) | 12 (13,6) | 10 (11,4) | 7 (7,9) | 2 (2,3) | 88 (100) | – |
| Edad promedio al inicio de la enfermedad en años (rango) | 8,2±3,9 de (2-14) | 6,2±3,3 de (0-13) | 8,0±3,5 de (2-13) | 6,3±4,4 de (1-13) | 5,9±4,8 de (0-14) | 4,5±2,1 de (3-6) | 7,1±3,8 de (0-14) | 0,213 |
| Distribución por género femenino: masculino | 3,1:1 | 2:1 | 2:1 | 4:1 | 1,3:1 | 2:0 | 2,1:1 | 0,5 |
| Intervalo promedio al diagnóstico en meses (mediana) | 11,9 (11) | 22,6 (11) | 13,6 (12) | 12,6 (12) | 23,9 (18) | 3,5 (4,5) | 16,5 (1-96) | 0,390 |
| Alteraciones estéticas n (%) | 25 (86,2) | 25 (89,3) | 12 (100) | 9 (90) | 7 (100) | 2 (100) | 80 (91) | 0,813 |
| Alteraciones funcionales articulares n (%) | 0 (0) | 15 (53,6) | 6 (50) | 6 (60) | 0 (0) | 1 (50) | 28 (32) | <0,001 |
| Alteraciones del crecimiento n (%) | 3 (10,3) | 17 (60,7) | 7 (58,3) | 2 (20) | 5 (71,4) | 2 (100) | 36 (41) | <0,001 |
La forma linear está dividida en compromiso en cara (CDS) y extremidades.
Fue la forma más común de esclerodermia. Correspondió a un tercio de los casos. El 51,7% (15/29) desarrolló lesiones múltiples. El área corporal más afectada fue el tronco en un 44,8% (13/29), seguido de miembros inferiores en 41,3% (12/29), cara en 17,2% (5/29), cuero cabelludo en 13,7% (4/29), miembros superiores en 13,7% (4/29) y cuello en 6,8% (2/29). Más del 80% de los pacientes presentaron discromía (82,7%) y un 6,8% presentó alopecia y efecto desfigurante. Ningún paciente presentó alteraciones funcionales articulares. El 6,8% (2/29) presentó alteraciones del crecimiento en miembros superiores (un paciente con compromiso longitudinal y un paciente con compromiso circunferencial). Un paciente presentó alteración del crecimiento en miembros inferiores (longitudinal). La morbilidad se relacionó, en su mayoría, con alteraciones estéticas dada la alta frecuencia de discromía y en menor proporción alopecia y efecto desfigurante (fig. 4).
Esclerodermia mixta
Una tercera parte de los pacientes desarrollaron formas mixtas de la enfermedad. El 75% (21/28) correspondió a la asociación entre morfea circunscrita y linear en extremidades. El 21,4% (6/28) asoció morfea circunscrita y CDS, y un paciente (3,6%) presentó la asociación de morfea circunscrita y lesiones lineares en extremidades y en cara (CDS).
Las áreas más comprometidas fueron el tronco y las extremidades inferiores en un 75% de los pacientes (21/28), seguidas de las extremidades superiores en 50% (14/28), cara en 32,1% (9/28), cuello en 14,2% (4/28) y cuero cabelludo en 3,5% (1/28). El 78% (22/28) presentó lesiones discrómicas, un tercio de los pacientes lesiones desfigurantes (9/28) y un 14,2% alopecia (4/28). Más de la mitad de los pacientes (53,6%) presentaron alteraciones funcionales en las articulaciones vecinas a la lesión. Se observaron alteraciones del crecimiento localizadas en extremidades inferiores en 13 pacientes (46,4%), siendo en 69% de los casos de compromiso mixto, en 23% longitudinal y en el restante circunferencial. En cara/cuero cabelludo la tasa de alteraciones del crecimiento fue de 28,5% (8/28) y en extremidades superiores de 14,2% (4/28), siendo en 75% de los casos de compromiso mixto y el restante circunferencial. Este tipo de esclerodermia se asoció a múltiples complicaciones tanto estéticas como funcionales, especialmente cuando presentaban lesiones lineares asociadas a lesiones de otro subtipo, lesiones múltiples o compromiso en cara.
Esclerodermia linearUn 21,5% (19/88) de los pacientes presentaron lesiones de distribución linear, pero si se incluyen los pacientes con formas mixtas, la frecuencia asciende al 55% de los casos (48/88). Siete pacientes presentaron la variedad en CDS y 12 pacientes presentaron lesiones lineares en extremidades.
Análisis descriptivo según subtipos de esclerodermia linear:
-Esclerodermia en CDS:
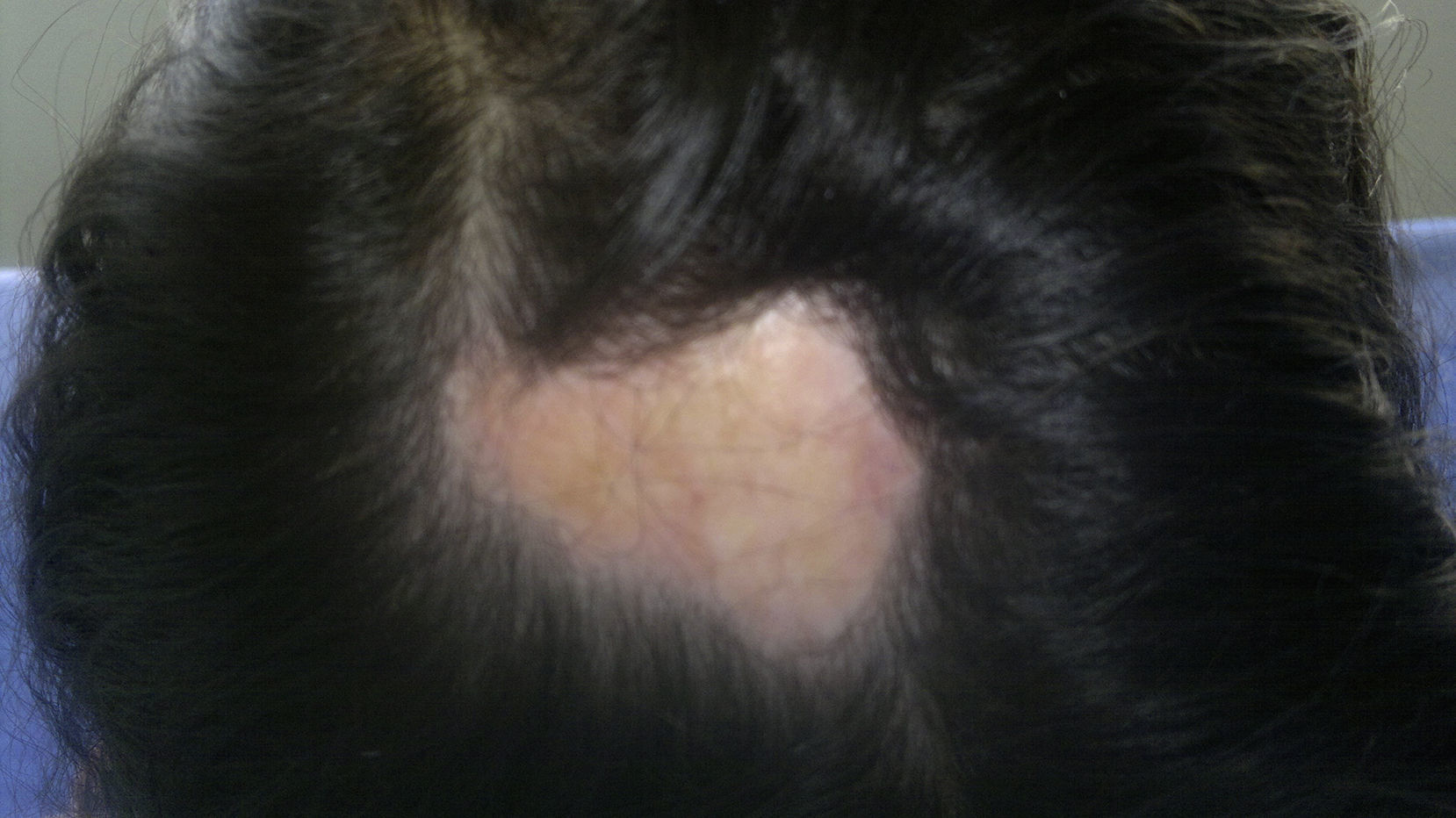
Además del compromiso en cara el 71,4% (5/7) asoció lesiones en cuero cabelludo. Lesiones múltiples estuvieron presentes en 42,8% (3/7) de los casos. Todos los pacientes desarrollaron alteraciones estéticas dadas por la presencia de lesiones discrómicas en 85,7% (6/7), lesiones desfigurantes en 85,7% (6/7) y un 57% adicional desarrolló alopecia localizada (4/7). Cinco de los pacientes (71,4%) presentaron alteraciones de crecimiento en cara/cráneo. Esta forma de esclerodermia presentó una importante tasa de alteraciones estéticas y alteraciones del crecimiento, así como el mayor tiempo de latencia entre el inicio de los síntomas y el diagnóstico.
-Esclerodermia linear en tronco y/o extremidades:
Las lesiones se ubicaron en igual proporción en extremidades superiores e inferiores (50% y 50%). No se presentó compromiso en tronco. La mayoría de los pacientes presentaron lesiones únicas (10/12, 83%). Todos los pacientes cursaron con alteraciones estéticas dadas por discromía. La mitad (6/12) cursó con compromiso funcional de articulaciones vecinas y con alteraciones del crecimiento en extremidades (3/12 en extremidades superiores y 3/12 en inferiores). El compromiso mixto fue el más frecuente en 4 de 6 pacientes con alteraciones del crecimiento, seguido del longitudinal. Uno de los pacientes requirió epifisiodesis de fémur distal y tibia proximal izquierdos para corrección de discrepancia de longitud en miembros inferiores y alargamiento del extensor del hálux y extensor común derecho por deformidad en garra del segundo artejo ipsilateral. La morbilidad de este subtipo de esclerodermia, además de las alteraciones estéticas, se asoció a una tasa importante de alteraciones del crecimiento (longitudinal, circunferencial o mixto) y compromiso funcional de articulaciones vecinas a la lesión.
Esclerodermia generalizadaTodos los pacientes con esta forma de esclerodermia presentaron compromiso en tronco y en miembros inferiores. El compromiso de miembros superiores se presentó en el 80% de los casos (8/10), en cuello en el 60% (6/10) y en cara en el 40% (4/10). No se presentaron lesiones en cuero cabelludo. El compromiso estético fue importante con la presencia de lesiones discrómicas en casi todos los pacientes (n=9, 90%) y efecto desfigurante en el 20% (n=2). Más de la mitad de los afectados desarrollaron compromiso funcional de articulaciones vecinas (n=6, 60%) y una quinta parte de los pacientes presentaron alteraciones del crecimiento (2/10, uno en cara y otro en extremidades superiores e inferiores con compromiso longitudinal). Un paciente presentó calcinosis. Se caracterizó por ser una forma con lesiones en piel extensas, importante tasa de alteraciones del crecimiento y fue la forma de esclerodermia con mayor frecuencia de compromiso funcional de articulaciones vecinas.
Esclerodermia panescleróticaSe presentó en 2 pacientes femeninas. Una paciente presentó compromiso articular dado por artritis y compromiso funcional poliarticular. Ambas presentaron alteraciones estéticas multifocales (extremidades, tronco y cara) y del crecimiento en extremidades (mixto). Una de ellas requirió amputación de la extremidad inferior izquierda secundaria a artritis séptica severa de rodilla. Se caracterizó por ser la forma más severa y progresiva que originó importante compromiso estético y funcional.
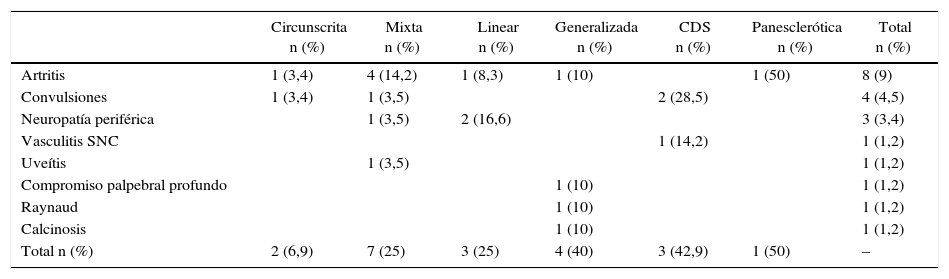
Compromiso extradérmicoSe detectó en una cuarta parte de los pacientes (n=20, 22,7%). El tipo con mayor frecuencia de compromiso extradérmico fue el CDS en 42,9% seguido de la esclerodermia generalizada en 40% de los pacientes. La forma linear fue la que con mayor frecuencia se asoció a compromiso articular (pura o dentro de formas mixtas). La afección del sistema nervioso central ocurrió con mayor frecuencia en pacientes con lesiones que comprometían cara/cráneo (tabla 2).
Compromiso extradérmico según tipos y subtipos de esclerodermia
| Circunscrita n (%) | Mixta n (%) | Linear n (%) | Generalizada n (%) | CDS n (%) | Panesclerótica n (%) | Total n (%) | |
|---|---|---|---|---|---|---|---|
| Artritis | 1 (3,4) | 4 (14,2) | 1 (8,3) | 1 (10) | 1 (50) | 8 (9) | |
| Convulsiones | 1 (3,4) | 1 (3,5) | 2 (28,5) | 4 (4,5) | |||
| Neuropatía periférica | 1 (3,5) | 2 (16,6) | 3 (3,4) | ||||
| Vasculitis SNC | 1 (14,2) | 1 (1,2) | |||||
| Uveítis | 1 (3,5) | 1 (1,2) | |||||
| Compromiso palpebral profundo | 1 (10) | 1 (1,2) | |||||
| Raynaud | 1 (10) | 1 (1,2) | |||||
| Calcinosis | 1 (10) | 1 (1,2) | |||||
| Total n (%) | 2 (6,9) | 7 (25) | 3 (25) | 4 (40) | 3 (42,9) | 1 (50) | – |
SNC: sistema nervioso central.
Once pacientes desarrollaron otra enfermedad autoinmune durante el seguimiento (12,5%). La más prevalente fue la artritis idiopática juvenil seguida de la tiroiditis de Hashimoto y el vitíligo. La distribución según subtipos de artritis idiopática juvenil fue 2 pacientes con la forma oligoarticular, uno con poliarticular y uno con artritis relacionada con entesitis. Dos pacientes presentaron enfermedad autoinmune múltiple definida como 3 o más enfermedades autoinmunes. Uno de estos con esclerodermia localizada, vitíligo y psoriasis, confirmados por biopsia en ese orden de aparición. Otro paciente asoció diabetes mellitus tipo 1, esclerodermia localizada y tiroiditis de Hashimoto, en ese orden de aparición (tabla 3).
Presencia de poliautoinmunidad según tipos y subtipos de esclerodermia
| Circunscrita n (%) | Mixta n (%) | Linear n (%) | Generalizada n (%) | CDS n (%) | Panesclerótica n (%) | Total n (%) | |
|---|---|---|---|---|---|---|---|
| AIJ | 2 | 1 | 1 | 4 (4,5) | |||
| Tiroiditis Hashimoto | 1 | 1* | 2 (2,3) | ||||
| Vitíligo | 1,1* | 2 (2,3) | |||||
| Lupus discoide | 1 | 1 (1,2) | |||||
| LES | 1 | 1 (1,2) | |||||
| Síndrome de Sjogren | 1 | 1 (1,2) | |||||
| Psoriasis | 1* | 1 (1,2) | |||||
| Diabetes mellitus tipo 1 | 1* | 1 (1,2) | |||||
| Total n (%) | 4 (13,7) | 4 (14,2) | 2 (16,6) | 1 (10) | 0 | 0 | – |
LES: lupus eritematoso sistémico.
En 46% de los pacientes se detectaron autoanticuerpos. En un 42% se detectaron anticuerpos antinucleares. ENAS fueron positivos en pacientes que asociaron LES o síndrome de Sjogren y en 2 pacientes sin otra enfermedad autoinmune asociada. Anti-DNA estuvo presente en el paciente con LES. Factor reumatoide se detectó en 2 pacientes, uno de ellos desarrolló artritis como compromiso extradérmico de la esclerodermia. Dos pacientes con tiroiditis de Hashimoto con Ac antitiroideos positivos. Dos pacientes con anti-SCL70 positivos. Anticardiolipina IgG y anticoagulante lúpico estuvieron presentes, cada uno en un paciente. Durante el tiempo de seguimiento ninguno de los pacientes desarrolló clínica o serología compatible con esclerosis sistémica.
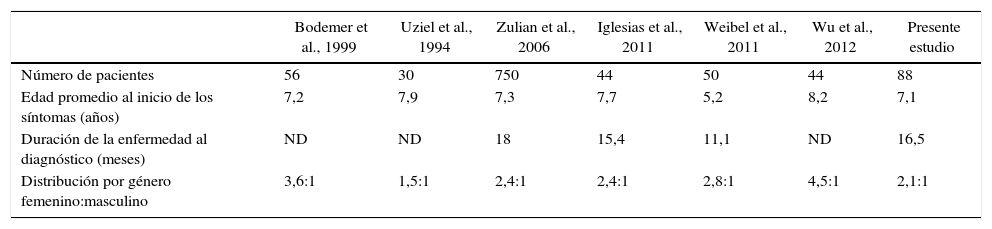
DiscusiónEste estudio representa la primera caracterización de un grupo de pacientes colombianos con jLS. Los estudios reportan una mayor frecuencia en el género femenino4-7 con una edad de inicio de síntomas alrededor de los 7 años (tabla 4), siendo coherentes con los hallazgos de este estudio4–6,8,11,13. El predominio en el género femenino fue más marcado en la esclerodermia generalizada, con una distribución por género femenino 4: masculino 1.
Cuadro comparativo entre diferentes series pediátricas de esclerodermia localizada
| Bodemer et al., 1999 | Uziel et al., 1994 | Zulian et al., 2006 | Iglesias et al., 2011 | Weibel et al., 2011 | Wu et al., 2012 | Presente estudio | |
|---|---|---|---|---|---|---|---|
| Número de pacientes | 56 | 30 | 750 | 44 | 50 | 44 | 88 |
| Edad promedio al inicio de los síntomas (años) | 7,2 | 7,9 | 7,3 | 7,7 | 5,2 | 8,2 | 7,1 |
| Duración de la enfermedad al diagnóstico (meses) | ND | ND | 18 | 15,4 | 11,1 | ND | 16,5 |
| Distribución por género femenino:masculino | 3,6:1 | 1,5:1 | 2,4:1 | 2,4:1 | 2,8:1 | 4,5:1 | 2,1:1 |
ND: no dato.
En las series de Zulian et al.4, Uziel et al.5 y Wu et al.13, se reporta el compromiso linear como el más frecuente, acorde con lo presentado en este estudio4,5,13. En relación a la distribución por tipos de esclerodermia, debe considerarse que dadas las diferencias en la clasificación de la entidad provoca dificultades para comparar hallazgos en los diferentes estudios.
A pesar de que la esclerodermia provoca lesiones cuyas características de brillo, induración y discromía deberían favorecer el diagnóstico temprano, se observa en todas las series una alta proporción de pacientes con diagnóstico tardío4–6,8,14,15 (tabla 4).
Resulta sorprendente que en esta serie, las formas mixtas y el CDS presentaron los mayores tiempos de latencia entre el inicio de los síntomas y el diagnóstico, pese a la multiplicidad de lesiones que caracterizan a las formas generalizadas y lo evidente de las lesiones en cara en el CDS. Esta demora en el diagnóstico puede estar relacionada con consultas tardías, falta de reconocimiento de la enfermedad por parte del médico o remisión tardía a dermatología y reumatología.
Las lesiones determinaron alteraciones estéticas en el 91% de los casos. Estas se clasificaron como discromía, alopecia y efecto desfigurante. Las lesiones de esclerodermia de los subtipos linear, mixta, generalizada y panesclerótica provocaron alteraciones funcionales de las articulaciones vecinas en más de la mitad de los casos.
En 58, 60, 71,4 y 100% de los pacientes con las formas linear, mixta, CDS y panesclerótica, respectivamente, se desarrollaron alteraciones localizadas del crecimiento. Uziel et al.5, reportan una tasa del 26% de alteraciones del crecimiento en pacientes con esclerodermia linear en extremidades5. Zulian et al.4, reportan atrofia de una extremidad con autoamputación en un paciente con la forma panesclerótica4 y Wu et al.13, reportan acortamiento de extremidades en 5 pacientes13. El presente estudio describe el compromiso del crecimiento en los distintos tipos y subtipos de esclerodermia.
Se ha reportado un mayor compromiso extradérmico en pacientes juveniles al comparar con adultos16. Gorkiewicz et al.17, reportan una tasa de compromiso extradérmico del 24 a 64% de los pacientes con esclerodermia localizada en adultos y niños (promedio del 20%), siendo mayor en las formas lineares y generalizadas17. Un 22,4% de los pacientes en la serie de Zulian et al.18, que incluyó exclusivamente pacientes juveniles (n=750), desarrolló compromiso extradérmico, con una frecuencia similar a lo observado en esta serie (22,7%)18.
La artritis fue la manifestación clínica del compromiso extradérmico más frecuente. Al igual que lo reportado por Zulian et al.18, esta ocurrió con mayor frecuencia en el compromiso linear de extremidades. El compromiso neurológico es el segundo más frecuente, caracterizado por convulsiones, neuropatía periférica y vasculitis de SNC. Este último paciente se caracterizó clínicamente por cefalea. Al igual que en la serie descrita el compromiso neurológico fue más prevalente en pacientes con lesiones en cara18.
El caso de uveítis se presentó en un paciente con CDS. La frecuencia de compromiso ocular fue menor a lo reportado en la literatura14,18. Esta diferencia puede estar determinada por un subdiagnóstico dado que no se realizó evaluación oftalmológica regular en todos los pacientes con lesiones en cara/cuero cabelludo.
El compromiso neurológico y ocular en pacientes con lesiones en cara/cuero cabelludo sugiere la necesidad de una evaluación neurológica y oftalmológica regular en estos pacientes.
El 12,5% presentó poliautoinmunidad. Esta tasa fue mayor al 7% reportado por Zulian et al.18, lo que sugiere la necesidad de elevar el nivel de alerta de aparición de otras enfermedades autoinmunes.
Al momento del seguimiento no ha habido progresión de la enfermedad a esclerosis sistémica en ningún paciente.
Dentro de las limitaciones del estudio se encuentran la ausencia de información acerca de antecedentes familiares de autoinmunidad, tratamiento recibido en los pacientes y que no se utilizó una herramienta estandarizada de seguimiento en los pacientes.
En conclusión, este estudio permite la caracterización de la jLS en un grupo de pacientes colombianos y demuestra que no se trata de una enfermedad limitada a la piel. A pesar de los avances en el tema la mayoría de las veces el diagnóstico es tardío. La esclerodermia circunscrita se acompaña de alteraciones estéticas importantes, mientras las formas linear, mixta, generalizada y panesclerótica se acompañan también de alteraciones funcionales importantes y alteraciones del crecimiento localizadas. La morbilidad asociada se acompaña de un impacto negativo y permanente en la calidad de vida de los pacientes con diagnóstico de jLS.
ConclusionesLa jLS es una enfermedad polimórfica e impredecible. Determina una morbilidad importante. El diagnóstico tardío es frecuente en adultos y en niños, y puede acompañarse de un impacto negativo en el pronóstico al permitir la progresión de las lesiones en tamaño, número y profundidad. El compromiso extradérmico se presentó casi en una cuarta parte de los pacientes. Esto refleja que no se trata de una enfermedad limitada a la piel.
La presencia de poliautoinmunidad se observó en una frecuencia no despreciable y en 2 pacientes se desarrolló enfermedad autoinmune múltiple. Se confirma la posible desregulación del sistema inmune como pieza importante en la patogénesis de la enfermedad. Un diagnóstico temprano, un tratamiento dinámico y un seguimiento cercano permiten prevenir y detectar tempranamente complicaciones derivadas de la enfermedad. Es necesario concientizar al personal médico acerca del carácter no benigno de esta condición. Se debe promover el aprendizaje, el reconocimiento temprano de esta patología y una remisión oportuna al reumatólogo. Así mismo, se debe garantizar el entendimiento por parte del paciente y cuidadores acerca de la enfermedad, la importancia de la adherencia al tratamiento y el seguimiento médico y la detección temprana de complicaciones con el objetivo de mejorar el pronóstico en los pacientes con diagnóstico de jLS.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
