



La granulomatosis con poliangeítis es un tipo de vasculitis que afecta a vasos de mediano y pequeño calibre de manera sistémica, con una alta expresión de anticuerpos contra el citoplasma del neutrófilo. Se presenta el caso de un paciente con un compromiso inicial de la vía área inferior, que no respondió al tratamiento y requirió manejo en unidad de cuidados intensivos. Finalmente, falleció por una hemorragia alveolar difusa severa. Su diagnóstico definitivo se estableció con una autopsia clínica. La granulomatosis con poliangeítis tiene diferentes formas de presentación y puede tener desenlaces fatales si no se diagnostica a tiempo.
Granulomatosis with polyangiitis is a systemic vasculitis that affects medium and small vessels, with high expression of anti-neutrophil cytoplasmic autoantibody. A case is presented on a patient with an initial compromise of the lower airway, who did not respond to management, required intensive care unit management, and died due to severe diffuse alveolar haemorrhage. His definitive diagnosis was established with a clinical autopsy. Granulomatosis with polyangiitis is a disease with different ways of presentation, and can have fatal outcomes if it is not diagnosed early.
La granulomatosis con poliangeítis (GPA) es una enfermedad sistémica compleja tipo vasculitis que afecta a vasos de mediano y pequeño calibre1. Se le conoce por el epónimo de enfermedad de Wegener, en honor a su primer descriptor, el patólogo alemán Friedrich Wegener2. Esta patología tiene una etiología compleja de tipo multifactorial, baja incidencia, al igual que su prevalencia, y se presenta de diferentes maneras. El diagnóstico habitualmente se realiza mediante una combinación de manifestaciones clínicas y pruebas de laboratorio, en tanto que el tratamiento depende de varios factores que influyen en el desenlace, así como en la progresión de la enfermedad3.
El presente artículo es un reporte de caso de un paciente masculino de 16 años con diagnóstico de GPA, con una presentación sistémica temprana y severa, que ingresó con una sintomatología sugestiva de neumonía asociada con expectoración hemoptoica, y no respondió a distintas líneas de manejo antimicrobiano durante su hospitalización, por lo que requirió manejo en unidad de cuidados intensivos (UCI) por falla ventilatoria. El diagnóstico definitivo de GPA se realizó con autopsia clínica a los 16 días del inicio de los síntomas.
El objetivo de este artículo es registrar y analizar el caso clínico de un paciente con GPA, el cual tuvo una presentación sistémica y temprana que resultó fulminante.
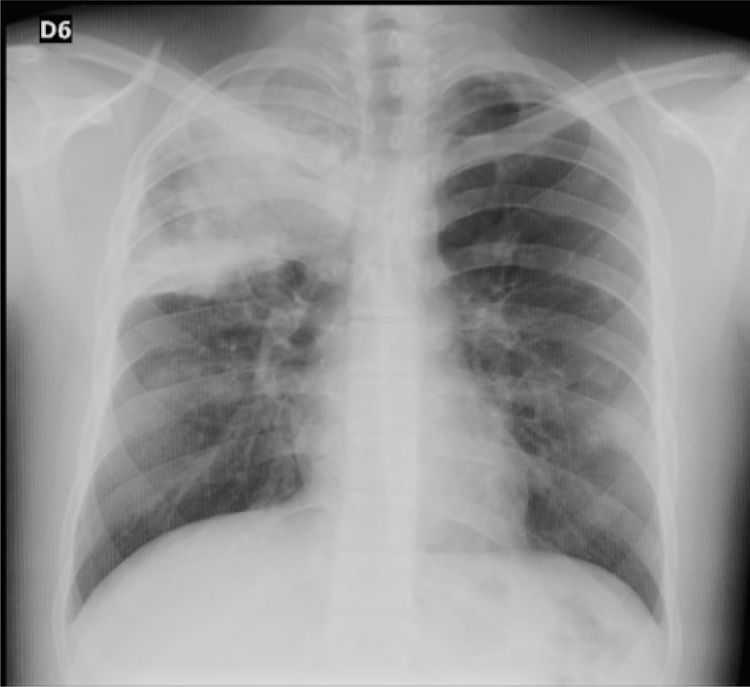
Reporte de casoPaciente varón de 16 años, con antecedentes de rinitis y conjuntivitis alérgica, hiperhidrosis manejada con simpatectomía bilateral y antecedente materno de hipotiroidismo, que consultó por sintomatología de 10 días de evolución de rinorrea hialina, congestión nasal, odinofagia, tos no productiva, malestar general, cefalea global, epistaxis y fiebre cuantificada en 39° C. Al ingreso presentó los siguientes valores de signos vitales: frecuencia cardiaca de 121, peso de 67kg, talla de 161cm, IMC de 25,8kg/m2, sin agregados pulmonares, con saturación de oxígeno de 95%, y sin otros hallazgos al examen físico. La radiografía de tórax mostró una consolidación en el lóbulo superior derecho (fig. 1), por lo cual se inició manejo para neumonía adquirida en la comunidad, de manera ambulatoria, con eritromicina oral durante 7 días.

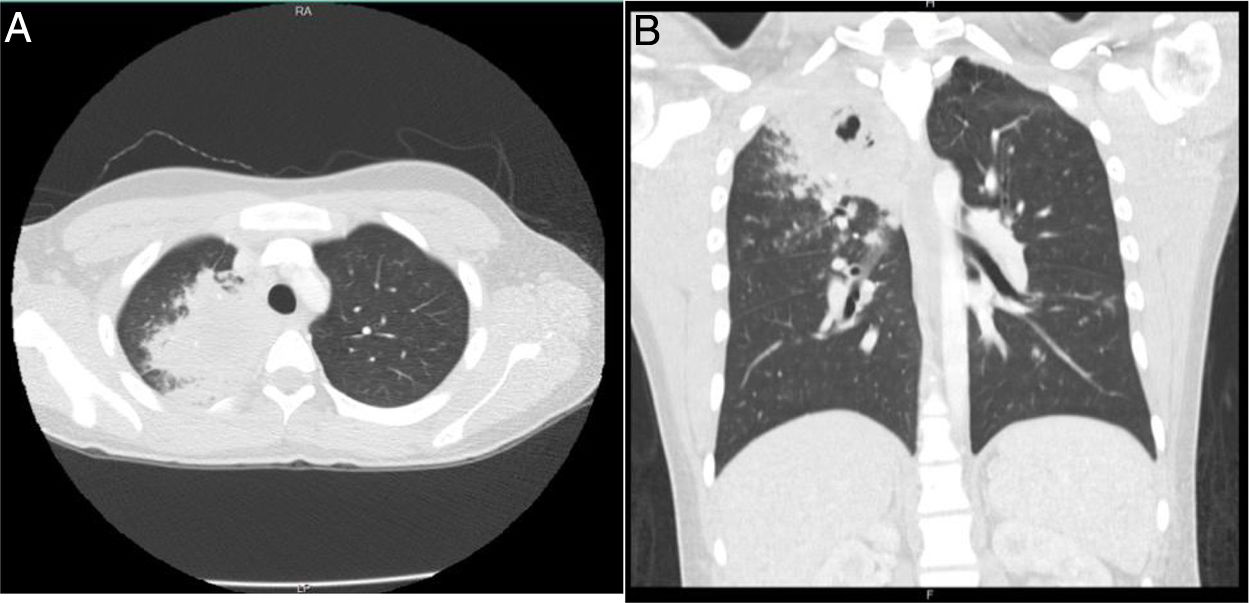
A las 24 h, el paciente reingresó por persistencia de la sintomatología, a la que se sumaron náuseas, vómito, dolor abdominal y disnea, por lo que fue manejado con broncodilatadores nebulizados, líquidos endovenosos, metoclopramida y dipirona intravenosa, con mejoría parcial de los síntomas. Después presentó deterioro clínico, con aparición de hemoptisis, deposiciones diarreicas fétidas, signos vitales con tensión arterial de 112/57mmHg, frecuencia cardiaca de 109, frecuencia respiratoria de 18, temperatura de 36,8° C, saturación de oxígeno de 96%, aparición de estertores finos en ápice derecho, sin lesiones en fosas nasales, leucocitosis importante (16.680), eosinofilia (11,4%) y bandas 9% (tabla 1). Debido a ello se hospitalizó y se inició manejo empírico con ampicilina sulbactam y claritromicina. La coloración de Gram y el cultivo de esputo fueron negativos, la tomografía de tórax mostró opacidades parenquimatosas que comprometían el segmento apical del lóbulo superior derecho e imágenes micronodulares que configuraban «árbol en gemación», con mínima necrosis central (fig. 2). Se tomaron nuevos hemocultivos y coprocultivo, los cuales resultaron negativos. El paciente presentó un nuevo pico febril prolongado de difícil manejo, por lo que se inició vancomicina. Se llevó a cabo lavado broncoalveolar por broncoscopia, en el cual el hidróxido de potasio, el bacilo de Koch y las bacterias tuvieron resultado negativo.
Datos de laboratorio
| Variables | Valores de referencia | Admisión | Día 5 | Día 6 | Día 8 | Día 11 | Día 14 |
|---|---|---|---|---|---|---|---|
| Hemograma | |||||||
| Leucocitos (células/uL) | 5.000-10.000 | 16.688 | 14.880 | 13.570 | 10.410 | 17.340 | 27.800 |
| Neutrófilos (células/uL) | 36,7-64,1 | 12.090 (72,5%) | 11.530 (77,5%) | 11.210 (82,6%) | 8.670 (83,3%) | 13.720 (79,1%) | 23.510 (84,6%) |
| Linfocitos (células/uL) | 21,2-39,7 | 1.600 (9,6%) | 880 (5,9%) | 750 (5,5%) | 1.150 (11%) | 2.180 (12,6%) | 3.070 (11%) |
| Monocitos (células/uL) | 4,05-12,8 | 1.050 (6,3%) | 1.050 (7,1%) | 610 (4,5%) | 560 (5,4%) | 870 (5,0%) | 970 (3,5%) |
| Eosinófilos (células/uL) | 1-3,9 | 1.900 (11,4%) | 1.370 (9,2%) | 960 (7,1%) | 0,0 (0%) | 540 (3,1%) | 220 (0,8%) |
| Basófilos (células/uL) | 0,01-1 | 40 (0,3%) | 50 (0,4%) | 40 (0,3%) | 30 (0,2%) | 30 (0,2%) | 30 (0,2%) |
| Bandas (%) | 0 | 9 | 3 | 0 | 0 | 0 | 0 |
| Hemoglobina (g/dl) | 14-18 | 11,9 | 10,5 | 12,1 | 11,3 | 10,1 | 7,8 |
| Hematocrito (%) | 45-56 | 38,5 | 33,9 | 38,1 | 34,2 | 30,7 | 23,5 |
| Volumen corpuscular medio (fl) | 80-100 | 83,9 | 82,9 | 82,1 | 80,7 | 81,6 | 82,5 |
| Hemoglobina corpuscular media (pg) | 27-34 | 25,9 | 25,7 | 26,1 | 26,7 | 26,9 | 27,4 |
| Plaquetas (células/uL) | 150.000-450.000 | 579.000 | 506.000 | 589.000 | 911.000 | 744.000 | 575.000 |
| Química sanguínea | |||||||
| Proteína C reactiva (mg/dl) | <6 | 12 | 48 | 24 | |||
| Procalcitonina (ng/ml) | 0-0,500 | 0,218 | 0,52 | 0,56 | |||
| Nitrógeno ureico (mg/dl) | 5,1-18 | 17,1 | 10 | 23,8 | 21,7 | ||
| Creatinina (mg/dl) | 0,67-1,17 | 0,78 | 0,85 | 0,77 | 1,17 | ||
| Transaminasa ALT/TGP (u/l) | <26 | 135,6 | 70,2 | ||||
| Transaminasa AST/TGO (u/l) | 26-31 | 49,1 | 24,1 | ||||
| Albúmina (g/dl) | 3,50-5,0 | 3,2 | 2,6 | ||||
| Fibrinógeno (mg/dl) | 0-339 | 462 | |||||
| Ácido úrico (mg/dl) | 2,1-7,6 | 4,6 | |||||
| Citoquímico de orina | |||||||
| Proteínas (mg/dL) | Negativo | Negativo | Negativo | 25 | 75 | ||
| Leucocito / estearasa | Negativo | Negativo | Negativo | Negativo | Negativo | ||
| Eritrocitos (celulas/ uL) | Negativo | 25 | 250 | 250 | 250 | ||
| Sedimento urinario | |||||||
| Hematíes (células x campo) | Negativo | 4 xC | >20 xC | >20 xC | >20 xC | ||
| Frescos | Negativo | 50% | 20% | 20% | |||
| Crenados | Negativo | 50% | 30% | 80% | |||
| Inmunológicos | |||||||
| Inmunoglobulina E (UI/ml) | 0-60 | 312 | 99,5 | ||||
| Inmunoglobulina A (UI/ml) | 90-310 | 105 | |||||
| Inmunoglobulina G (UI/ml) | 710-1.520 | 975 | |||||
| Inmunoglobulina M (UI/ml) | 40-157 | 73 | |||||
| Complemento C3 (mg/dl) | 90-180 | 175,1 | |||||
| Complemento C4 (mg/dl) | 10,0-40 | 28,1 | |||||
| ANCA-MPO (U/ml) | 11,0-18 | 0,6 | |||||
| ANCA-PR3 (U/ml) | 11,0-18 | 238,2 | |||||
ALT/TGP: alanino amino tranferasa/transaminasa glutámico pirúvica; ANCA: anticuerpo contra el citoplasma del neutrófilo; ANCA-MPO: ANCA contra la mieloperoxidasa; ANCA-PR3: ANCA contra la proteinasa 3; AST/TGO: aspartato amino transferasa/transaminasa glutámico oxalacética.
 corte axial. B) Corte coronal. Opacidad parenquimatosa en el segmento apical del lóbulo superior derecho, con áreas sugestivas de cavitación con imágenes micronodulares, lo que configura un «árbol en gemación».")
A los 5 días el paciente presentó inadecuada respuesta al manejo, se solicitó complemento que mostró C3 aumentado, con ANCA-PR3 positiva (anticuerpo contra el citoplasma del neutrófilo-proteinasa 3), y en el hemograma se detectó persistencia de la leucocitosis (14.880) y eosinofilia (9,2%). Se realizó prueba cutánea de tuberculina (PPD) que salió negativa, una tomografía de senos paranasales normal y nuevos hemocultivos que resultaron negativos. Se sospechó vasculitis granulomatosa eosinofílica vs. granulomatosis con poliangeítis (enfermedad de Wegener); se inició corticoide endovenoso y se modificó el manejo antibiótico a ceftriaxona con clindamicina. El paciente presentó un nuevo pico febril, se escaló el antibiótico nuevamente a vancomicina y se continuó con ceftriaxona. Los hemocultivos de control fueron negativos, persistió la leucocitosis (17.000) y se inició con trombocitosis (744.000). El paciente presentó dolor abdominal epigástrico y episodios eméticos, por lo que se inició omeprazol, sin que se registrara mejoría, y se tomó ecografía abdominal que mostró hepatomegalia y alteración difusa de la ecogenicidad renal, pero con función renal conservada.
A los 16 días de su admisión, el paciente presentó mal estado general, deshidratación grado 2, palidez generalizada, frecuencia cardiaca de 135 y saturación de oxígeno del 92% con FiO2 al 50%. Asimismo, estaba polipneico, con tirajes supraclaviculares, disminución de murmullo vesicular, descenso en la hemoglobina (7,8) y leucocitosis franca (27.800). Una nueva radiografía de tórax mostró infiltrados alveolares generalizados de predominio de formación periférica, por lo cual se consideró que cursaba con una hemorragia alveolar difusa vs. síndrome de dificultad respiratoria del adulto, por lo que se trasladó a la UCI. Ya en la unidad, el paciente presentó deterioro en su patrón respiratorio, saturación de oxígeno del 82% con FiO2 al 50%, accesos de tos severos con hemoptisis franca, y persistió con dificultad respiratoria, por lo que se realizó intubación orotraqueal y se inició ventilación mecánica. El uroanálisis mostró proteinuria y hematuria, y los tiempos de coagulación eran los normales; también presentaba hipoproteinemia e hipoalbuminemia. Se transfundieron glóbulos rojos empaquetados, se realizó panel viral y otra baciloscopia y hemocultivo, los cuales fueron negativos. El ecocardiograma transtorácico mostró hipertensión pulmonar moderada a severa (psap=72mmHg), con hipertrofia del ventrículo derecho y disfunción, por lo cual se inició manejo con dobutamina, noradrenalina y ciclofosfamida intravenoso con pulsos de corticoide. Se realizó una nueva fibrobroncoscopia y se puso en evidencia una hemoptisis moderada. El paciente persistió con deterioro, por lo que se adicionó manejo con milrinona, sin mejoría, y se inició plasmaféresis. Además, presentó síndrome pulmón/riñón, con falla renal no oligúrica y hemorragia alveolar difusa severa. Se inició reposición de plasma y albúmina, pero el paciente no respondió al manejo y persistió con trastorno de la oxigenación.
A los 3 días de su ingreso en la UCI, el paciente presentó bradicardia progresiva con hipotensión, sin respuesta a vasopresores a dosis máximas. Persistió con desaturación marcada (40%) con máximo soporte ventilatorio, sucesivamente presentó 3 episodios de paro cardiaco, los cuales respondieron a manejo con adrenalina y compresiones torácicas. En el último evento presentó asistolia que no respondió al manejo, se realizó reanimación cardiopulmonar avanzada, pero no se logró la presencia de signos vitales, por lo cual se declaró el fallecimiento.
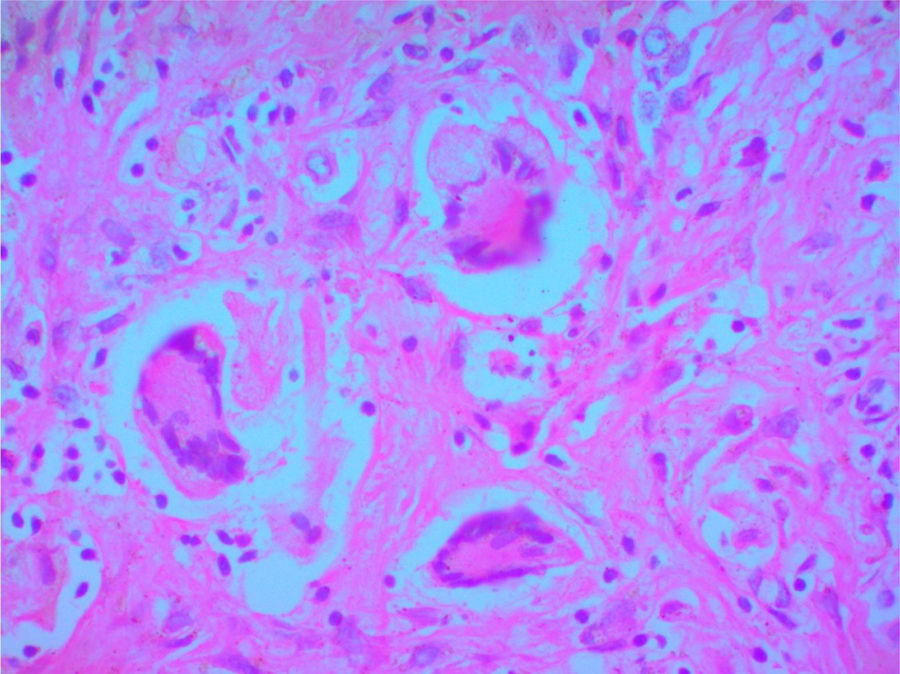
Se solicitó autopsia clínica por causa no clara del deceso, y se encontró a nivel macroscópico, en el lóbulo superior derecho del pulmón, una lesión irregular con induración y área de necrosis en la superficie. En el parénquima se observaron múltiples lesiones nodulares y en el resto del parénquima pulmonar un aspecto hemorrágico. A nivel microscópico, el parénquima pulmonar presentó hemorragias alveolares y zonas de necrosis (fig. 3), además de granulomas rodeados por una zona de proliferación fibroblástica con infiltrado leucocitario y células gigantes multinucleadas con vasos obliterados por vasculitis. Se realizaron coloraciones especiales de Periodic Acid-Schiff, Ziehl-Neelsen y Gromory, las cuales fueron negativas para microorganismos y positivas para fibrinógeno en espacios alveolares. Estos hallazgos confirmaron el diagnóstico de GPA (granulomatosis de Wegener).
Discusión
La GPA es una enfermedad sistémica asociada a ANCA, definida por el consenso internacional de Chapell Hill como una vasculitis necrotizante con afectación de vasos de pequeño a mediano calibre, asociada con una inflamación granulomatosa que puede comprometer la vía aérea superior e inferior y el riñón, con glomerulonefritis y necrosis focal4. Esta patología tiene una incidencia estimada en Norteamérica de 10,8 casos nuevos por millón de personas/año (IC 95%: 10,7-12,9)5, y en Sudamérica de 9 casos por millón personas/año, con una prevalencia de 7,4 por cada 100.000 habitantes (IC 95%: 2,8-12)6. Su presentación en la población pediátrica es mayor en mujeres (70%)7; sin embargo, en nuestro caso se presentó en un hombre joven, de 16 años, sin factores de riesgo ni antecedentes importantes.
Desde el inicio, las manifestaciones clínicas presentadas correspondieron a un compromiso de la vía aérea inferior, con una discreta afectación de la vía aérea superior (rinorrea hialina)8, lo que demuestra la heterogeneidad en su presentación, ya que habitualmente el compromiso reportado en estudios clínicos consiste en afectación renal, síntomas constitucionales (fiebre, pérdida de peso, mialgias, entre otros) y manifestaciones otorrinolaringológicas9. En un primer momento, la mala respuesta al uso de antimicrobianos debió orientar a que no se trataba de una patología infecciosa. Múltiples hemocultivos, cultivos directos de lavados broncoalveolares y muestras de esputo, no aislaron ningún germen. Sin embargo, se ha determinado que infecciones por bacterias como el Estafilococo aureus pueden sugerir una posible etiología de esta vasculitis10. Existen otros estudios que describen factores de riesgo diferentes, como la disminución en la exposición a rayos ultravioleta o una mayor latitud, que podrían predisponer a presentar esta enfermedad11, al igual que el antecedente de uso de ciertos medicamentos como los antitiroideos o los antibióticos puede favorecer la producción de ANCA por linfocitos B policlonales12.
En cuanto al manejo recibido, el inicio de corticoide intravenoso pudo condicionar la respuesta al tratamiento, obteniéndose una meseta en la progresión de la enfermedad, ya que se encuentra dentro de las líneas de manejo propuestas por la Liga Europea contra el Reumatismo (EULAR, por sus siglas en inglés), pero no se observó respuesta alguna cuando se usó ciclofosfamida en bolos, ni tampoco algún cambio cuando se realizó plasmaféresis13. Se contempló el uso de rituximab, un anticuerpo monoclonal contra el antígeno CD-20 de los linfocitos B14, pero la inadecuada respuesta y la presencia de hemorragia alveolar difusa acortaron su sobrevida, por lo cual no se pudo intentar el manejo con este medicamento.
Al presentarse unas manifestaciones clínicas heterogéneas, mala evolución clínica y pobre respuesta al manejo farmacológico, con desenlace fatal para el paciente, se decidió hacer una confirmación de este diagnóstico con una autopsia clínica, ya que la presencia de fiebre durante la hospitalización impidió que se le realizara una biopsia pulmonar para confirmar la GPA. Finalmente, el reporte de patología demostró la existencia de granulomas con zonas de necrosis en espacios alveolares, lo que demostró que efectivamente era una GPA, pero con una presentación sistémica temprana fulminante.
ConclusionesLa GPA es una patología compleja de tipo multifactorial, con diversas manifestaciones clínicas, y no se han unificado criterios específicos para poder emitir un diagnóstico precoz e inicio temprano de tratamiento. En este momento se está llevando a cabo el estudio Diagnostic and Classification Criteria in Vasculitis, un estudio observacional que unificará estos criterios y permitirá un adecuado manejo temprano de los pacientes15. Las formas de presentación tempranas y severas influyen en el pronóstico de la enfermedad, y por ende en su mortalidad, por lo que se requieren ensayos clínicos que indaguen en este tipo de presentación16. Se encuentra en vigor el tratamiento sugerido por la EULAR, y el seguimiento debe realizarse de conformidad con los lineamientos propuestos14. El objetivo en el futuro para la medicina actual será realizar una identificación oportuna y temprana de este tipo de casos, para poder iniciar un manejo precoz de la enfermedad y lograr un impacto importante en la supervivencia y mejorar así su pronóstico.
Consideraciones éticasEl caso cuenta con consentimiento informado en el cual se autoriza al uso de la información y divulgación de los datos clínicos del paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen el apoyo del Departamento de Patología de la Universidad de La Sabana, en especial al doctor Néstor Segundo Beleño Beltrán y a la doctora María Claudia Abaúnza Chaguin, por la digitalización de las imágenes obtenidas.
