




APS is a hypercoagulability condition characterized by the development of thrombosis and pregnancy morbidity (recurrent early miscarriages, fetal deaths after the 10th week of gestation and/or premature births), that occur in patients with antiphospholipid antibodies, namely lupus anticoagulant, anticardiolipin antibodies, and anti-β2-glycoprotein-I antibodies. It is usually isolated but can occur in the setting of another autoimmune disease, mainly systemic lupus erythematosus. Moreover antiphospholipid antibodies can be found in individuals without the disease. Treatment of thrombosis is based on indefinite anticoagulation while low-dose aspirin and low molecular weight heparin are the cornerstone of pregnancy morbidity treatment. Catastrophic antiphospholipid syndrome is treated with anticoagulation, plasma-exchange, and corticosteroids. Standardization of serological assays, inclusion of other antibodies and manifestations in the classification criteria, treatment of non-criteria manifestations and refractory cases are areas of uncertainty.
El SAF es una condición de hipercoagulabilidad caracterizada por el desarrollo de trombosis y morbilidad obstétrica (abortos recurrentes, muertes fetales antes de la semana 10 de gestación y/o partos prematuros) en pacientes con anticuerpos antifosfolipídicos, específicamente el anticoagulante lúpico, los anticuerpos anticardiolipina y anti-β2-glicoproteína-1. En la mayoría de los casos se presenta de forma aislada, pero puede asociarse a otras enfermedades autoinmunes como el lupus eritematoso sistémico. Además, los anticuerpos antifosfolipídicos se pueden encontrar en individuos sin la enfermedad. El tratamiento de la trombosis se basa en anticoagulación indefinida, mientras que aspirina a dosis bajas y heparina de bajo peso molecular representan la base del tratamiento de la morbilidad obstétrica. El síndrome de anticuerpos antifosfolipídicos catastrófico se trata con una combinación de anticoagulación, corticoides y recambios plasmáticos. La estandarización de los ensayos serológicos, la inclusión de otros anticuerpos y otras manifestaciones en los criterios clasificatorios, el tratamiento de las manifestaciones no criterio y de los casos refractarios representan las áreas de incertidumbre del síndrome.
When August Von Wasserman developed his test for the diagnosis of syphilis,1 based on an auto-antibody (called “reagin”) directed against an antigen from lipoid tissue, which was later purified and named cardiolipin by Mary C Pangborn,2 he could never imagine that he was laying the first stone toward the discovery of a syndrome that would have been described, eventually, by Graham Hughes, almost 80 years later.3 Since its first description, advances in recognition of both the clinical and pathophysiological aspects of the condition have been notable, and even though antiphospholipid syndrome (APS) was originally described as an acquired autoimmune thrombophilia, we know that other mechanisms are involved in several manifestations of the disease.
APS is a hypercoagulability condition characterized by the development of arterial, venous and/or microvascular thrombosis, and pregnancy morbidity (recurrent early miscarriages, fetal deaths after the 10th week of gestation and/or premature births), that occur in patients with persistent antiphospholipid antibodies (aPL) namely lupus anticoagulant (LAC), IgG or IgM anticardiolipin antibodies (aCL), or IgG or IgM anti-β2-glycoprotein-I antibodies (aβ2GPI). APS can occur either as an isolated condition (primary APS), or in the context of an underlying autoimmune disease, most commonly systemic lupus erythematosus (SLE). Less frequently, it can be associated with other autoimmune conditions, infections, drugs and malignancies.
The original description of the syndrome was made by Graham Hughes in 1983,3 even though the first reports of thrombosis in patients with SLE and LAC date back to late 1950s and early 1960s.4–6 Single vessel involvement or multiple vascular occlusions may give rise to a wide variety of presentations in the APS. Any combination of vascular occlusive events may occur in the same individual and the time interval between them also varies considerably from weeks to months or even years. The “Euro-Phospholipid” project, a study of 1000 European APS patients,7 has provided accurate information on the prevalence of the majority of clinical manifestations of this syndrome, which is now recognized as a major cause of deep vein thrombosis (DVT) with or without pulmonary embolism, new strokes in individuals below the age of 50 and recurrent fetal loss. The major nonthrombotic manifestations include livedo reticularis, valvular heart disease, APS-related nephropathy, chorea, epilepsy, memory loss, migraine and myelopathy. Hematologic alterations, such as hemolytic anemia and thrombocytopenia are also very common. In a subset of patients (about 1%), thrombosis can involve simultaneously multiple organs, configuring the so-called “catastrophic antiphospholipid syndrome” (CAPS).8 This review highlights the epidemiology, pathogenesis and the most common clinical manifestations as well as the management of this autoimmune disease.
EpidemiologyThe aPL are not specific of APS and can be found in healthy individuals. Nevertheless, the prevalence of aPL positivity and APS in the general population has not been extensively analyzed and only two epidemiological population-based studies have been performed so far. In the first one, the authors studied the epidemiology of APS between 2000 and 2015 in an inception cohort of Olmsted County, Minesota, through a record linkage system. The annual incidence of APS in adults aged ≥18 years was 2.1 (95% confidence interval 1.4–2.8) per 100,000 population. Incidence rates were similar in both sexes. The estimated prevalence of APS was 50 (95% CI 42–58) per 100.000 population, and was similar in both sexes.9 In the second study, performed in Korea between 2007 and 2018, with data extracted from the Health Insurance and Review Agency, an incidence of 0.75 per 100,000 person-year (95% confidence interval 0.73–0.78) was found, while the prevalence was 6.19 per 100,000 people.10
The prevalence of DVT occurrence in the general population is estimated at 2–5%, 10–20% associated with APS, suggesting that the prevalence of venous thrombosis associated with APS may be as high as 0.3–1% of the general population.11 Moreover, the prevalence of aPL has been estimated about 11% among patients with myocardial infarction and 17% among patients with stroke younger than 50 years of age.12 aPL antibodies are present in 30–40% of SLE patients and up to a third of these patients (10–15% of SLE patients) have clinical manifestations of APS, especially venous or arterial thromboses.13,14 On the contrary, only few patients with primary APS tend to evolve into full-blown SLE and, usually, this takes place only after a long period of time.15 Among women with pregnancy complications, the prevalence of aPL is about 6%, and aPL are now regarded as the most frequent acquired risk factor for a treatable cause of recurrent pregnancy loss and for pregnancy complications (early and severe pre-eclampsia).12,13 The prevalence of CAPS has been estimated to be less than 1% of all APS patients.16
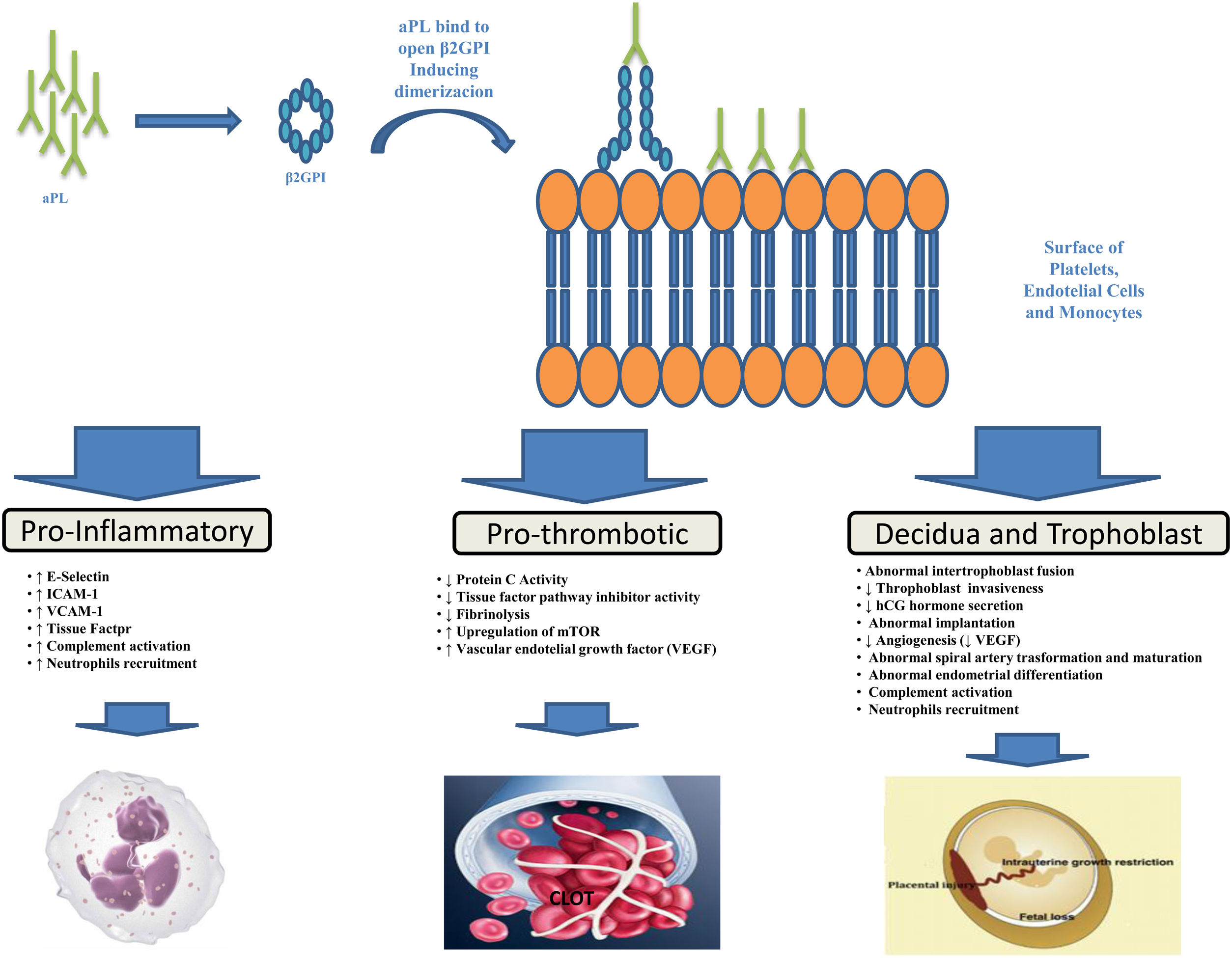
PathogenesisThe aPL are heterogeneous antibodies and more than one mechanism may be involved in causing thrombosis. As demonstrated by various studies, the major target of aPL is β2-glycoprotein I (β2GPI), a plasma protein that binds avidly to phospholipid surfaces, whose binding with aβ2GPI leads to its conformational change and dimerization (the immunogenic form of β2GPI).17–21 The binding of aPL to β2GPI on the surfaces of platelets, endothelial cells and monocytes up-regulates the expression of prothrombotic cellular adhesion molecules such as E-selectin, ICAM-1, VCAM-1,22 and of tissue factor23 suppressing the activity of the tissue factor pathway inhibitor,24 reducing activated protein C activity,25 and activating complement.26 Annexin A2,27 a tissue plasminogen activator receptor, toll like receptor-428,29 and apoE-receptor-230 may serve as intermediary. A possible explanation for microvascular thrombosis in APS is the aPL-induced up-regulation of the mechanistic target of rapamycin (mTOR) complex on endothelial cells.31
Pregnancy morbidity was initially related to the impairment of maternal-fetal blood exchange as a result of thrombus formation in the uteroplacental vasculature, an hypothesis supported by findings of placental thrombosis in patients with obstetric APS.32 However, such a histologic finding is not specific for APS, being also present in other conditions, and histologic evidence of thrombosis in the uteroplacental circulation cannot be shown in many placentas from patients with APS. Other theories have thus been put forward to explain APS-related pregnancy morbidity such as defective trophoblast invasion33 and decidual transformation in early pregnancy or placental injury as a result of local inflammatory events, particularly complement activation and neutrophils recruitment.32,34 The function of complement seems particularly interesting in such setting and a prospective, multicenter, observational study entitled PROMISSE (Predictors of Pregnancy Outcome: Biomarkers in Antiphospholipid Antibody Syndrome and SLE – NCT00198068) to examine the role of complement as a potential surrogate marker that predicts poor pregnancy outcomes in patients with APS is under way and scheduled for completion in 2021. Figure 1 provides a brief summary of the pathophysiological mechanisms leading to thrombosis and pregnancy morbidity in APS.
 produced by B cells bind to open, immunogenic, β2-glycoprotein I (β2GPI), leading to conformational change and dimerization. Annexine A2, Toll Like receptor-4 and apoE-receptor-2 may serve as receptor for β2GPI on cell surfaces. This binding results in endothelial-cell, monocyte, platelet and neutropphil activation and trophoblast and decidua modification leading to inflammation, thrombosis and pregnancy complications.")
Antiphospholypid antibodies (aPL) produced by B cells bind to open, immunogenic, β2-glycoprotein I (β2GPI), leading to conformational change and dimerization. Annexine A2, Toll Like receptor-4 and apoE-receptor-2 may serve as receptor for β2GPI on cell surfaces. This binding results in endothelial-cell, monocyte, platelet and neutropphil activation and trophoblast and decidua modification leading to inflammation, thrombosis and pregnancy complications.
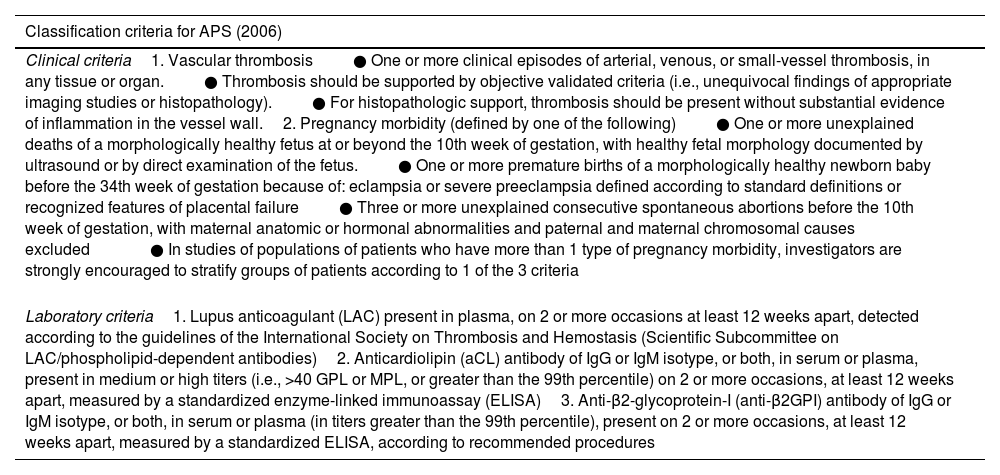
According to the present classification criteria of APS, stated in 2006 during the 11th International Congress on Antiphospholipid Autoantibodies,35 diagnosis can be made in the presence of at least one clinical manifestation (either thrombosis or pregnancy morbidity) along with the positivity (at medium-high titer) of one or more aPL in at least two occasions 12 weeks apart (Table 1). The aCL and anti-β2GPI are detected via solid-phase immunoassays (usually ELISAs),36 while LAC test is performed following the Scientific and Standardization Subcommittee on Antiphospholipid Antibodies of the International Society of Thrombosis and Haemostasis (SSC-ISTH) recommendations.37 For instance, LAC is detected through a three-step procedure which involves prolongation of phospholipid-dependent clotting time such as diluted Russell viper venom time (dRVVT) and the activated partial thromboplastin time (aPTT) not reversed mixing patient plasma with normal plasma, but reversed by the addition of excess phospholipids. One of the major drawbacks of the LAC coagulation assays is that they can be altered by anticoagulant therapy, giving false-positive results.37 Furthermore, the aCL and aβ2GPI antibodies assays show interassay variation owing to differences in calibration and differences in assay characteristics.38
Adapted from Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306; with permission.
| Classification criteria for APS (2006) |
|---|
| Clinical criteria1. Vascular thrombosis● One or more clinical episodes of arterial, venous, or small-vessel thrombosis, in any tissue or organ.● Thrombosis should be supported by objective validated criteria (i.e., unequivocal findings of appropriate imaging studies or histopathology).● For histopathologic support, thrombosis should be present without substantial evidence of inflammation in the vessel wall.2. Pregnancy morbidity (defined by one of the following)● One or more unexplained deaths of a morphologically healthy fetus at or beyond the 10th week of gestation, with healthy fetal morphology documented by ultrasound or by direct examination of the fetus.● One or more premature births of a morphologically healthy newborn baby before the 34th week of gestation because of: eclampsia or severe preeclampsia defined according to standard definitions or recognized features of placental failure● Three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes excluded● In studies of populations of patients who have more than 1 type of pregnancy morbidity, investigators are strongly encouraged to stratify groups of patients according to 1 of the 3 criteria |
| Laboratory criteria1. Lupus anticoagulant (LAC) present in plasma, on 2 or more occasions at least 12 weeks apart, detected according to the guidelines of the International Society on Thrombosis and Hemostasis (Scientific Subcommittee on LAC/phospholipid-dependent antibodies)2. Anticardiolipin (aCL) antibody of IgG or IgM isotype, or both, in serum or plasma, present in medium or high titers (i.e., >40 GPL or MPL, or greater than the 99th percentile) on 2 or more occasions, at least 12 weeks apart, measured by a standardized enzyme-linked immunoassay (ELISA)3. Anti-β2-glycoprotein-I (anti-β2GPI) antibody of IgG or IgM isotype, or both, in serum or plasma (in titers greater than the 99th percentile), present on 2 or more occasions, at least 12 weeks apart, measured by a standardized ELISA, according to recommended procedures |
Abbreviations: GPL, G phospholipid units; MPL, M phospholipid units.
Since aPL can be present in healthy individuals and in a majority of conditions (such as infections, neoplasms and other autoimmune diseases), a generalized search for aPL in the absence of any relevant condition is strongly discouraged to prevent incidental findings. APS must be suspected in case of a young patient presenting with unprovoked thrombosis, especially if at unusual sites and recurrent, or in thrombotic or pregnancy complications associated to other autoimmune diseases. Venous thromboembolism is the most frequent manifestation in APS, with a frequency of 39% in the Europhospholipid Project cohort.7 Patients with venous thromboembolism most commonly present with lower-extremity DVT, pulmonary embolism, or both. Stroke and transient ischemic attack are the most common arterial events. Combined, DVT (usually in the legs) and ischemic stroke account for 90% of all complications.39 The following accompanying clinical findings may be a clue that a patient has APS: unexplained prolongation of the aPTT, livedo reticularis or racemosa, signs or symptoms of another systemic autoimmune disease, and mild thrombocytopenia. Severe thrombocytopenia (platelet count, <20,000 per cubic millimeter) is rare40 and should prompt the clinician to consider other causes. Thrombosis recurrence is a hallmark of APS; interestingly, patients with arterial thrombosis have a higher risk of recurrence compared with those with venous thrombosis, and a tendency for recurrences in the same vascular (arterial) bed is the rule.41 Other risk factors for recurrence are triple aPL positivity, LAC persistent positivity, and associated SLE.42
Recurrent miscarriages at <10 weeks of gestation are the most frequent obstetric manifestation of APS.43 However, the most typical complications of pregnancy generally develop after 10 weeks of gestation and losses before 10 weeks, especially if not recurrent, would more commonly be attributed to chromosomal defects (which must always be excluded to make a diagnosis). Late pregnancy loss, with early or severe preeclampsia, or with the HELLP syndrome (hemolysis, elevated liver-enzyme levels, and low platelet counts) are the typical obstetric manifestations. Reduced blood flow in the uterine arteries measured by Doppler velocimetry is an indirect indicator for the development of placental insufficiency, intrauterine growth restriction and/or preeclampsia.44,45 Thenceforth, the European League Against Rheumatism (EULAR) guidelines recommend the use of uterine artery Doppler ultrasonography during pregnancy monitoring.46
The major nonthrombotic manifestations are hemolytic anemia, thrombocytopenia, livedo reticularis (a reddish-blue to purple, uniform, reversible, unbroken “net-like” pattern of the skin), livedo racemosa (nonuniform, irreversible, fractured, asymmetric pattern), livedoid vasculopathy (painful papules and erythematous-violaceous, purpuric plaques, which rapidly evolve into hemorrhagic vesicles or painful small ulcers), valvular heart disease, pulmonary hypertension, diffuse alveolar hemorrhage, APS-related nephropathy (acute or chronic thrombotic microangiopathy), adrenal hemorrage, chorea, epilepsy, memory loss and cognitive disfunction (due to aPL related vasculopathy or direct aPL interactions with brain parenchyma following blood-brain barrier abrogation), migraine, and myelopathy.7
Catastrophic APS is a rare, life-threatening form of APS that occurs in less than 1% of patients and is characterized by involvement simultaneously or in less than a week, of multiple organs, tissues or systems. It usually follows a precipitating factor, such as infection (in almost half of cases), anticoagulation withdrawal, neoplasm, surgery or pregnancy. Histological confirmation of small vessel occlusion is necessary to make a diagnosis as per classification criteria.47
Sometimes a high clinical suspicion of APS is not supported by concomitant positivity of aPL assays included in the serological criteria for APS (LAC and IgG and IgM isotypes of aCL and aβ2GPI antibodies) which are persistently negative. This is the framework of the so-called seronegative APS which has been described by Hughes and Khamashta in 2003.48 Thenceforth, numerous investigators looked for the presence in these patients of aPL not included in the serological criteria for APS. For instance, these non-criteria antibodies include aCL and aβ2GPI IgA, antibodies specific to phospholipid-binding plasma (cofactor) proteins (such as phosphatidylethanolamine, prothrombin, protein C, protein S, annexin V and domain I of β2GPI), phospholipid–protein complexes (particularly vimentin–cardiolipin complexes), and anionic phospholipids other than cardiolipin (including phosphatidylserine, phosphatidylinositol and phosphatidic acid).49–54 In case of highly suspected APS with persistently negative LAC, aCL and aβ2GPI IgG and IgM, after ruling out other causes of thrombophilia, looking for these non-criteria antibodies can suggest the diagnosis.
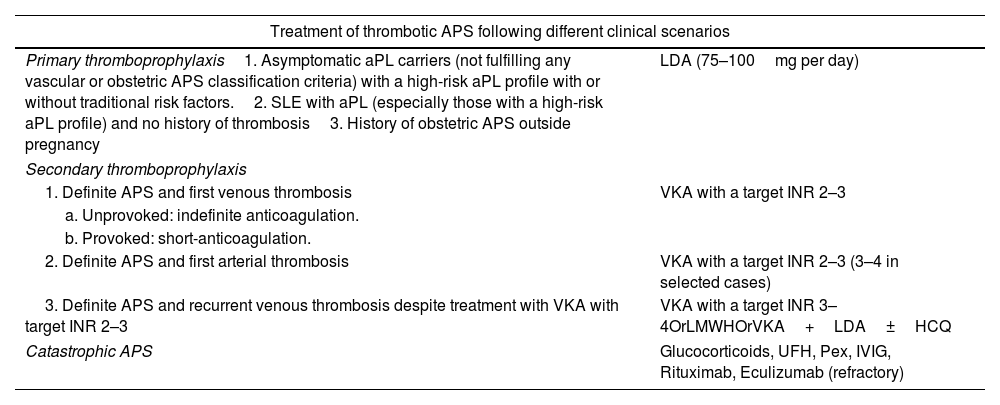
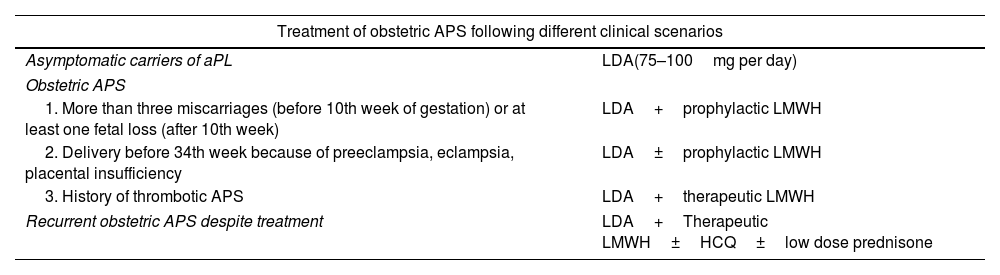
TreatmentThe management of APS has been subject to controversy in recent years. Anticoagulation therapy is considered the cornerstone of treatment, but the optimal agents and the intensity of treatment remain a matter of debate. Recently, updated guidelines on the treatment of APS by the EULAR have been published.55 However, since APS is a fairly new and rare disease, good-quality data to guide treatment are scarce and treatment decisions rely on expert opinion in many cases. The treatment of APS varies depending of the clinical manifestations, aPL profile, and concurrent cardiovascular risk factors. Treatment options in different clinical scenarios are reported in Tables 2 and 3.
| Treatment of thrombotic APS following different clinical scenarios | |
|---|---|
| Primary thromboprophylaxis1. Asymptomatic aPL carriers (not fulfilling any vascular or obstetric APS classification criteria) with a high-risk aPL profile with or without traditional risk factors.2. SLE with aPL (especially those with a high-risk aPL profile) and no history of thrombosis3. History of obstetric APS outside pregnancy | LDA (75–100mg per day) |
| Secondary thromboprophylaxis | |
| 1. Definite APS and first venous thrombosis | VKA with a target INR 2–3 |
| a. Unprovoked: indefinite anticoagulation. | |
| b. Provoked: short-anticoagulation. | |
| 2. Definite APS and first arterial thrombosis | VKA with a target INR 2–3 (3–4 in selected cases) |
| 3. Definite APS and recurrent venous thrombosis despite treatment with VKA with target INR 2–3 | VKA with a target INR 3–4OrLMWHOrVKA+LDA±HCQ |
| Catastrophic APS | Glucocorticoids, UFH, Pex, IVIG, Rituximab, Eculizumab (refractory) |
Abbreviations: aPL, antiphospholipid antibodies; APS, antiphospholipid syndrome; LDA, low dose aspirin; SLE, Systemic Lupus Erithematosus; VKA, Vitamin K antagonist (e.g. Warfarin); INR, international normalized ratio; LMWH, low molecular weight heparin (at therapeutic dose); UFH, unfractioned heparin; Pex, plasma exchange; IVIG, intravenous immunoglobulin.
High risk profile: the presence (in 2 or more occasions at least 12 weeks apart) of lupus anticoagulant (measured according to ISTH guidelines), or of double (any combination of lupus anticoagulant, anticardiolipin (aCL) antibodies or anti-β2-glycoprotein-I antibodies) or triple (all three subtypes) aPL positivity, or the presence of persistently high aPL titers.
Low risk profile: isolated aCL or anti-β2-glycoprotein-I antibodies at low-medium titers, particularly if transiently positive.
| Treatment of obstetric APS following different clinical scenarios | |
|---|---|
| Asymptomatic carriers of aPL | LDA(75–100mg per day) |
| Obstetric APS | |
| 1. More than three miscarriages (before 10th week of gestation) or at least one fetal loss (after 10th week) | LDA+prophylactic LMWH |
| 2. Delivery before 34th week because of preeclampsia, eclampsia, placental insufficiency | LDA±prophylactic LMWH |
| 3. History of thrombotic APS | LDA+therapeutic LMWH |
| Recurrent obstetric APS despite treatment | LDA+Therapeutic LMWH±HCQ±low dose prednisone |
Abbreviations: aPL, antiphospholipid antibodies; APS, antiphospholipid syndrome; LDA, low dose aspirin; LMWH, low molecular weight heparin; HCQ, hydroxychloroquine.
LDA must be started before conception. LMWH must be continued up to 6 weeks after delivery.
It is not infrequent that a patient is found to be positive for aPL during an evaluation for a systemic autoimmune disease or because of an elevated activated partial-thromboplastin time (aPTT), or a false positive result of syphilis test. In such cases it must be considered that aPL represent a risk factor for thrombosis and pregnancy complications, which are commonly multifactorial. Thus, a risk stratification based on age, aPL profile, concomitant genetic and acquired risk factors for thrombosis (such as dyslipidemia, smoke, hypertension, diabetes, contraceptive use, menopause, etc.) along with the presence of systemic autoimmune diseases must be taken into accountjhh and a strict follow-up is mandatory. A major risk factor is the high-risk aPL profile, including any of the following: the presence of LAC as the aPL subtype most closely related to thrombosis,56 the presence of double (any combination of LAC, aCL and aβ2GPI antibodies) or triple aPL positivity, or the presence of persistently high (above 40 IgG or IgM phospholipid units or >99th percentile) aCL or aβ2GPI titers.57 Furthermore, thrombosis is more strongly associated with IgG isotype than with the IgM isotype antibodies.58 A score that takes into accounts cardiovascular risk factors (namely hypercholesterolemia and hypertension) and the aPL profile, the Global Anti-Phospholipid Syndrome Score (GAPSS), has shown to be related to thrombotic and obstetric events probability.59,60
The use of low-dose aspirin (LDA) for primary thrombosis prevention is controversial since the quality of evidence is low.61 The APLASA trial,62 that studied primary thromboprophylaxis with LDA in asymptomatic aPL carriers, did not show efficacy, but it was underpowered to detect any difference between LDA and placebo. A meta-analysis of seven observational studies of 460 asymptomatic aPL carriers found the risk of first thrombosis to be reduced by half in those who used LDA versus those who did not use LDA.63 Therefore, the last EULAR recommendations suggest to treat aPL carriers with a high-risk profile and/or a concomitant SLE or patients with obstetric APS outside pregnancy with LDA.55 A moderate-to-high-risk aPL profile warrants avoidance of estrogen-based contraceptives when possible and aggressive postoperative prophylaxis with low molecular weight heparin (LMWH) if feasible.
In patients with venous thrombosis related to APS, after an initial therapy with unfractionated or LMWH, a long-term anticoagulant therapy with a vitamin K antagonist such as warfarin (target international normalized ratio [INR] 2–3), is recommended. Higher intensity anticoagulation, with a target INR 3 to 4, did not further reduce the risk of recurrent thrombosis, in two randomized clinical trials.64,65 Indefinite anticoagulation in patients with unprovoked venous thromboembolism is highly warranted, due to the high risk of thrombosis recurrence in case of VKA discontinuation.66 Nevertheless, in case of provoked first venous thrombosis (as after surgery, prolonged immobility, long-distance travel, etc.), the benefit of long-term anticoagulation is less clear, and therapy should be discontinued – especially in cases with transient positivity and low-risk aPL profile – as in patients without APS, according to international guidelines.67
In case of arterial thrombosis, treatment with VKA with a target INR of 2–3 has showed no difference in thrombosis recurrence compared to a target of 3–4 in two clinical trials.64,65 Nevertheless, the higher intensity INR approach is preferred by some centers, due to the low number of patients with arterial thrombosis included in the aforementioned trials. The association of VKA and LDA is often reserved to patients with clinically significant risk factors for cardiovascular disease or patients in whom a single antithrombotic agent has failed to prevent recurrence.57 In decision-making, physicians should take into account the individual's risk of recurrent thrombosis and major bleeding, as well as the patient's preferences after discussion.
In case of thrombosis recurrence, high-quality evidence to support any particular management strategy when warfarin therapy fails despite a target INR is lacking. Viable options include higher intensity warfarin therapy (target INR, 3–4), switch to LMWH, the addition of LDA, antimalarials,68 statins, or a combination of these approaches.55
Since the introduction on the market of direct oral anticoagulants (DOACs) in 2010, they received increasing attention due to the obvious advantages in terms of quality of life for patients who have to follow a long-term, often lifetime, VKA treatment and have to come every 2–3 weeks to the clinic to get an INR determination. The recent TRAPS trial analyzed the efficacy of rivaroxaban, a direct factor X inhibitor, in comparison with warfarin for prevention of thrombosis recurrence in triple aPL positive patients with previous arterial thrombosis, showing an excess of arterial thrombosis in patients on rivaroxaban. Therefore, DOACs are not recommended in patients with arterial thrombosis.69
Prevention of pregnancy complications in asymptomatic patients with aPL, especially those with high risk profile, is based on LDA (75–100mg per day), even though good evidence is lacking.55 Pregnant women with previous obstetric APS should be treated with a combination of LDA and a prophylactic dose of unfractionated or LMWH,70 with a live birth rate of about 75%,71 even though the quality of evidence is low.72 LDA should be preferably started prior to conception, and heparin should be added as soon as pregnancy is confirmed. LMWH is preferred for practical reasons. Oral anticoagulants should be discontinued at conception because of teratogenicity between 6 and 14 weeks of gestation. Heparin should be continued up to 6 weeks after delivery to prevent maternal thrombosis, given the increased thromboembolic risk in puerperium. In case of recurrent pregnancy morbidity despite combination therapy, increasing heparin dose to therapeutic dose, addition of hydroxychloroquine73 or low-dose prednisolone74 in the first trimester may be considered. Intravenous immunoglobulins are an option in refractory cases,75 albeit results are contradictory.76 Even though statins are not typically used in pregnancy, a case-control study which analyzed the use of pravastatin with standard of care in APS patients with pre-eclampsia and/or intrauterine growth restriction showed no progression compared to LDA and LMWH.77 The putative mechanism of action has been investigated in a very recent study and it seems to be increased nitric oxide synthesis.78 In women with a history of thrombotic APS, a combination treatment of LDA and heparin at therapeutic dosage during pregnancy is recommended, regardless of obstetric history.
Since long term risk of thrombosis for women with obstetrical APS is lower than the risk for women whose syndrome-defining event was thrombotic,79 long-term antithrombotic therapy for women who have a history of obstetrical APS but no other risk factors for thrombosis is not recommended.
A prompt and aggressive treatment is critical in case of catastrophic APS, and the current standard of care is the so-called triple therapy, a combination of anticoagulants, glucocorticoids, and plasma exchange.80 Intravenous immunoglobulins (1–2g/kg, given over a period of 2–5 days) are often associated to the triple therapy and, as well as rituximab, are an option for refractory cases.81 Complement inhibition (e.g. eculizumab) may also be an option for refractory cases.81 Given the rarity of the syndrome, non-controlled studies have been done, and the proposed therapies are based on low-quality evidence.
Finally, the so-called non-criteria manifestations represent a gray area of treatment guidelines. Thrombocytopenia is pretty exclusively mild-to-moderate and does not require medical treatment. In the rare case of severe thrombocytopenia (platelets below 20,000 per cubic millimeter), treatment is based on glucocorticoids with or without intravenous immunoglobulins if indicated.82 Splenectomy is not a first-line treatment because of the increased risk of thrombosis for patients with the APS who undergo surgery.57 Second-line therapies include mycophenolate mofetil, cyclophosphamide, and azathioprine with evidence coming from case series and observational studies.82 Rituximab83 and thrombopoietin receptor agonists84 are indicated in refractory cases. First-line treatment for autoimmune hemolytic anemia in APS consists of high-dose corticosteroids, while traditional immunosuppressants, rituximab, or splenectomy have been used with varying success as second-line treatments in refractory cases.85
Evidence-based recommendations for the management of heart valve disease in APS are lacking. An earlier consensus report concluded that oral anticoagulation does not halt the development or progression of valve lesions, while prophylactic LDA may be considered in asymptomatic aPL-positive individuals with valve disease.85 Anticoagulation is recommended in patients with thromboembolic episodes attributed to valve disease and can be considered in case of vegetations due to the increased risk of thromboembolic stroke.86
There is no consensus about the treatment of neurologic manifestations associated to APS. Various case reports showed efficacy of antiplatelet and anticoagulant treatment, while the role of conventional immunosuppression is not clear.87 Case reports showed successful treatment of aPL-associated chorea with hydroxychloroquine, mycophenolate mofetil or intravenous immunoglobulins, but prospective studies are needed to examine their efficacy.
APS-related nephropathy is usually slowly progressive, histologically characterized by fibrous intimal hyperplasia, fibrocellular arterial occlusion, focal cortical atrophy and tubular thyroidization, and has no standard treatment. Anticoagulation is indicated in case of history of thrombotic APS, but its role in the evolution of renal function is unknown, owing to the limited number of patients and limited follow-up period in the majority of case series.85 Acute renal failure is typically associated with thrombotic microangiopathy and can be treated with rituximab,83 eculizumab88 and plasma exchange.89 In any case of aPL-associated nephropathy, strict control of arterial hypertension and proteinuria with angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers is highly recommended.
Not specific treatments are usually needed for livedo reticularis or livedo racemosa. Livedoid vasculopathy is usually refractory to glucocorticoids; LDA, dipyridamole, clopidogrel, pentoxifylline, sildenafil, intravenous immunoglobulins, tissue plasminogen activator, hyperbaric oxygen therapy, or a combination of these interventions, with or without anticoagulant therapy, have been used.57,90
Sometimes, patients who initially tested positive for aPL can become persistently negative. In such cases the question arises of whether it is possible to withdraw anticoagulant treatment. Coloma-Bazzan et al.91 described a series of 11 patients who presented no new thrombotic episode during a 20 month-follow-up after withdrawal of anticoagulation, suggesting that anticoagulation can be safely withdrawn in selected patients. However, discontinuation of VKA treatment in patients who became persistently negative to aPL needs further evidence.
Unmet needsAll assays routinely used to detect aPL show methodological shortcomings and lack of standardization. Harmonization of working conditions using automated systems may contribute to a reduction in interlaboratory variation92 and validation of several non-criteria antibodies assays, such as prothrombin, phosphatidylserine–prothrombin complex, domain 1, phosphatidic acid, annexin A5, aCL and aβ2GPI IgA.49
Since the current classification criteria do not incorporate the full spectrum of clinical findings for the APS, an international effort is under way to develop a more comprehensive classification, with the use of the same methods that were used to develop the most recent classification criteria for SLE.93
There are several areas of uncertainty in the management of APS in which evidence is scarce or nonexistent, such as treatment of non-criteria manifestations, seronegative APS and refractory cases of thrombotic and obstetric APS.
Treatment of obstetric APS with current standard of care results in live-birth rates above 70%, which means that about 30% of women continue to have pregnancy complications. A multicenter randomized controlled trial of hydroxychloroquine (associated to standard of care) versus placebo to improve pregnancy outcome in women with aPL (HYPATIA)94 is ongoing and results are awaited.
Last but not least, the possibility of withdrawal of anticoagulation in selected cases of thrombotic APS in which assays for aPL become persistently negative is another gray area where evidence is scarce, and further studies are warranted.
FundingNone declared.
Conflict of interestNone declared.
