



Desde los primeros estudios en inmunología, ha sido evidente la necesidad de entender cómo, en condiciones normales, el sistema inmune tolera los antígenos propios y ataca algunos antígenos extraños que percibe como potencialmente peligrosos, y cómo, bajo ciertas circunstancias, la pérdida de la tolerancia desencadena enfermedades autoinmunes. Ha pasado más de medio siglo desde que Billingham, Medawar y Brent demostraron en un modelo experimental algunos eventos involucrados en el desarrollo de la tolerancia inmunológica. Desde entonces, los inmunólogos de trasplante han centrado sus esfuerzos en dilucidar los mecanismos que conllevan al mantenimiento de la tolerancia, con la esperanza de eludir las complicaciones de la inmunosupresión no específica y conseguir la prevención del rechazo crónico.
Medawar (1953) argumentaba que durante el trasplante el sistema inmune del individuo se hacía tolerante al tejido trasplantado, manteniéndose la respuesta a otros antígenos. Estudios recientes han demostrado que la pérdida de la tolerancia al trasplante está asociada con una hiperrespuesta a los antígenos del tejido trasplantado, hecho que ha atormentado a los inmunólogos clínicos, quienes han encaminado sus esfuerzos a desarrollar sistemas de medición precisos que les permita evaluar qué tan tolerante podría ser un individuo al trasplante.
Los intentos por inducir tolerancia en el individuo, se basan en la comprensión de los mecanismos básicos de tolerancia, cuyo conocimiento se ha desarrollado paralelamente con una mejor apreciación de la complejidad de la tolerancia inmune. En particular, se ha avanzado mucho en la comprensión del papel esencial de las células dendríticas tolerogénicas (CDT) y del mantenimiento de la tolerancia por células T reguladoras.
Since the first studies in immunology, there has been a clear need to understand how, under normal conditions, the immune system tolerates its own antigens and attacks some foreign antigens that it perceives as potentially dangerous and how, in certain circumstances, the loss of tolerance triggers autoimmune diseases. It has been over half a century since Billingham, Brent and Medawar demonstrated, in an experimental model, the mechanisms involved in the development of immunological tolerance. Since then transplant immunologists have intensively investigated the mechanisms involved in maintaining tolerance, in the hope of avoiding the complications of non-specific immunosuppression, as well as the prevention of chronic rejection.
An important characteristic was observed by Medawar, who argued that during transplantation an individual's immune system is tolerant to transplanted tissue, maintaining the response to other antigens. Recent studies have shown that loss of tolerance to transplantation is associated with a hyper-response to antigens of the transplanted tissue; a problem that has plagued clinical immunologists, who have focused their efforts on developing accurate measurement systems to enable them to measure how an individual could be tolerant to transplant.
Attempts to induce tolerance in the individual are based on understanding the basic mechanisms of tolerance, in which there has been significant progress. This growth in knowledge has been in parallel with a better appreciation of the complexity of immune tolerance. In particular, progress has been made in understanding the essential role of tolerogenic dendritic cells (CDS) and the maintenance of tolerance by regulatory T cells.
Una característica distintiva del sistema inmune es la capacidad para discriminar entre lo propio y lo extraño, y de mantener la tolerancia frente a antígenos propios, así como de generar una respuesta inmune eficaz contra patógenos y células malignas. La relación entre la tolerancia y la inmunidad es dinámica y se conceptualiza en el término “dicotomía del sistema inmune”. La pérdida de dicha tolerancia desencadena eventos adversos que conducen a manifestaciones fisiopatológicas tales como: infecciones, tumores malignos o autoinmunidad1,2.
Con el fin de evitar eventos adversos perjudiciales para el organismo como autorreactividad, las células T y B, respectivamente, logran desarrollar autotolerancia durante estadios tempranos en el proceso de maduración intratímico y en la médula ósea3,4. El repertorio de células T se logra gracias al proceso de selección intratímica durante los mecanismos de tolerancia central, en el cual son eliminadas la mayoría las células T que desarrollan una alta autorreactividad4–7. Después de salir del timo, las células T maduras son sometidas a selección secundaria (tolerancia periférica), por lo que la mayoría de las células T autorreactivas son suprimidas o adquieren anergia8,9. En el riguroso proceso de selección, la regulación de la tolerancia central y periférica juega un papel importante; esto debido a la falta de coestimulación por células presentadoras de antígenos o por defectos en la expresión de marcadores de superficie. Además de estos mecanismos pasivos la evidencia, acumulada en los últimos 15 años, indica que las células T reguladoras (Tregs) CD4+CD25+, juegan un papel relevante en el mantenimiento de la autotolerancia periférica, así como en la regulación de las diversas reacciones inmunes10–12.
La autotolerancia periférica es esencial para el correcto funcionamiento del sistema inmune, pero también puede ser utilizada como un mecanismo de evasión mediante el cual las células tumorales evitan el reconocimiento y la destrucción por el sistema inmunológico. La interrupción de la autotolerancia por infecciones u otros mecanismos (anomalía genética o factores ambientales) contribuye al desarrollo de enfermedades autoinmunes; por lo tanto, es concebible pensar que el diseño de estrategias terapéuticas, a través de mecanismos de prevención o reversión de la tolerancia inmunológica, pueden ser utilizadas para mejorar la lucha contra este proceso, así como ayudar en el control de tumores, neoplasias malignas avanzadas, o en la supresión de autoinmunidad y respuestas inmunes no deseadas13,14.
La evidencia de modelos animales ha demostrado que la terapia con células tolerogénicas puede prevenir o curar el rechazo de trasplantes o enfermedades autoinmunes y que el papel de las células T reguladoras es esencial en la modulación de la respuesta inmunológica13.
La presente revisión hace un recorrido de los trabajos sobre tolerancia inmune, desde los trabajos de Born (1897) hasta los de Sakagushi (2002, 2004, 2008), Morelli (2007) y Matsumoto (2011), en los que se involucran aspectos importantes que evidencian el desarrollo de la tolerancia como son la dilucidación de los mecanismos de tolerancia central y periférica, desarrollados por los linfocitos T y B en el timo y en la médula ósea; así como el papel de las células T reguladoras y células dendríticas con actividad tolerogénica.
Recuento históricoLa definición de “tolerancia inmunológica” ha evolucionado lentamente después de la observación de R.D. Owen (1897) en terneros gemelos dicigóticos, en los que se observaba un intercambio constante de sangre embrionaria, que volvía tolerantes a los antígenos tisulares de ambos animales entre sí15. Lo anterior refutaba la creencia predominante de la época, según la cual las células de individuos diferentes eran rechazadas por la respuesta inmune, concepto que persistió durante mucho tiempo. Burnet y Fenner tomaron nota de estas observaciones y plasmaron una teoría que esclarecía los mecanismos involucrados en el control y en el rechazo, y la denominaron respuesta inmune adaptativa16. Burnet y Fenner basaron su teoría en la siguiente argumentación:
- 1.
El reconocimiento de lo extraño requiere de un reconocimiento de lo propio por parte del organismo.
- 2.
Este mecanismo de control se activa en la vida temprana.
- 3.
La evidencia podría apoyar este pensamiento inductivo.
Los mencionados autores argüían que si las células vivas de un ratón de la cepa CBA, se inyectaban en un ratón adulto de la cepa A, las células CBA serían destruidas por el sistema inmune, y el ratón de la línea A que recibía las células destruiría cualquier injerto posterior del mismo origen, con la velocidad que se esperaba de un animal inmunológicamente competente; pero si las células CBA se inyectaban en un feto o en un ratón recién nacido de una cepa A, las células serían aceptadas. Cuando estos individuos crecieran aceptarían cualquier injerto de un donante CBA, como si se tratara de sus propias células.
Medawar (1960) observó que sus experimentos se pueden considerar como “una reproducción artificial de una asombrosa curiosidad natural”. El y sus colegas demostraron que el intercambio de injertos de piel entre los gemelos dicigóticos quiméricos de vacunos era aceptado sin que se generara rechazo. Es importante señalar que en esa época no se contaba con métodos de medición precisos como los que dispone hoy en día la biología molecular. Sin embargo, Medawar, con marcada precisión, llegó a la conclusión que los antígenos se encontraban presentes en los tejidos, aunque en bajas dosis, y mantenían un umbral bajo de respuesta, sin que se generara autorreactividad17.
A raíz de los descubrimientos de Medawar, muchos estudios se han centrado en descifrar los mecanismos implicados en el desarrollo de la tolerancia, asociados principalmente con las dosis de antígenos presentes en los tejidos. Medawar argumentaba que las dosis de antígeno presentes en el órgano trasplantado podrían clasificar el nivel de tolerancia en alto y bajo. Un amplio grupo de investigadores se ha centrado en estudiar los antígenos asociados a tejidos in vivo y su papel en el desarrollo de la tolerancia. No se ha planteado en relación con la tolerancia inmunológica una hipótesis concreta que argumente que la eliminación de los clones autorreactivos se produce durante la vida fetal. Los antígenos extraños que están presentes durante este proceso putativo, no generan respuesta en la vida adulta. En otras palabras, el cuerpo en su fase embrionaria es engañado, pero cuando adquiere madurez, reacciona eliminando los antígenos extraños. Nossal (1969) en una revisión reflexiva, referencia este hecho al que Burnet y Fenner18 denominan “la discriminación entre el yo y el no-yo”.
También se ha llegado a creer que los mecanismos de tolerogénesis implican una incapacidad específica para responder a los antígenos y, tal vez, la supresión de células autorreactivas pueda ser uno de los temas de estudio en el futuro para explicar sus causas.
La tolerancia inmunológica a antígenos propios y no propios ha llegado a ser aceptada como parte de un mecanismo natural que, bajo ciertas circunstancias, impide la destrucción del propio cuerpo. También se ha llegado a creer que el desarrollo de las enfermedades autoinmunes se debe a alguna falla en los mecanismos tolerogénicos.
Evidencias embriológicasA pesar de la importancia de la respuesta inmune en el rechazo al trasplante, ella sólo comienza a tener relevancia con los trabajos realizados por Born en 1897, quien manifestaba que la inmadurez en las especies era una variable importante en la aceptación del trasplante. La inmadurez inmunológica en los embriones de anfibios y en los embriones de aves, era la causa de que algunos trasplantes se toleraran en la edad adulta. Born realizó trasplantes xenogénicos, creando quimeras, mediante la utilización de tejidos de rana trasplantados en embriones o larvas de sapos. Diez años más tarde, Lewis utilizó el modelo de Born, en tritones con el fin de estudiar la diferenciación de las vesículas ópticas, completando con éxito su diferenciación19. En 1934 Hewitt observó anormalidades en la diferenciación al realizar xenotrasplantes de vesículas ópticas, encontrando atrofia progresiva del injerto20.
Unos años antes de la Primera Guerra Mundial, Rous y Murphy, en 1911, utilizaron por primera vez embriones de pollo como reservorio de tejidos extraños. Inocularon embriones con células del sarcoma de rata logrando mantener vivos los huevos durante el periodo de incubación. Murphy (1911) demostró en los años siguientes que el “embrión de pollo no generaba defensas contra las células del sarcoma de rata”, que un pequeño infiltrado de células linfoides rondaba el tejido injertado y que la respuesta se generaba en el momento de la eclosión. Sin embargo, un descubrimiento de mayor trascendencia fue el realizado por el mismo Murphy (1914) con el cual demostró que el crecimiento in vitro del sarcoma de Rous es inhibido por la presencia de fragmentos de bazo o por células de la médula ósea de pollos adultos, mientras que en otros tejidos no tenían tal efecto21,22. Demostró, así mismo, que los embriones de aves no son inmunocompetentes y que su inmadurez puede ser revertida administrando células de adultos jóvenes inmunológicamente competentes23.
El trasplante de esbozos de miembros y otros tejidos se convirtió en un evento muy utilizado por los embriólogos experimentales después de la descripción en Hamburgo, en 1933, en la cual se demostró que el trasplante era una técnica relativamente simple. Años más tarde, Eastlick (1941), fue el primero en utilizar el término “tolerancia”24,25, trasplantó tejidos embrionarios de otras especies de aves en el embrión de pollo, muchos injertos sobrevivieron y se diferenciaron. Señaló que el tiempo de aparición y la severidad de las reacciones de incompatibilidad dependían de la especie donante24,26,27. En los experimentos realizados con injertos de pato se obtuvo un resultado inesperado, ya que uno de los injertos sobrevivió durante 13 meses, pero la mayoría no logró sobrevivir a la reacción que se desarrolló cerca o después de la eclosión. La reacción afectó seriamente el sistema vascular del injerto, indicado por la estasis y el edema. Aunque no se ha demostrado experimentalmente, parece lógico suponer que las proteínas extrañas del injerto sensibilizaron al huésped poniendo en juego los mecanismos de defensa.
A propósito de lo anterior, Eastlick (1941) especuló que entre el injerto embrionario y el tejido del huésped del experimento pudo haberse presentado una disminución del “ajuste”, de lo que dedujo que la tolerancia no se puede perder completamente; este fue el concepto más próximo a la compresión del evento al que se pudo llegar en la época para entender el éxito en los trasplantes. Lo anterior se asociaba con la idea de que la presencia de tejido extraño en el desarrollo del embrión generaba un cambio en la respuesta del huésped: hay pérdida relativa de la tolerancia, no hay el suficiente ajuste y, por lo tanto, se rechaza el injerto.
A comienzos del siglo xx, Paul Ehrlich habló del “horror autotoxicus” para referirse a las lesiones causadas por la activación inadecuada del sistema inmune contra tejidos propios. Desde entonces, los estudios sobre los mecanismos involucrados en el mantenimiento de la tolerancia han contribuido de manera importante al desarrollo de la inmunología de los trasplantes. Los trabajos desarrollados por Owen, Medawar, Burnet y otros, durante los años 40 y 50, demostraron que la tolerancia a lo propio se desarrollaba durante la vida embrionaria y sentaron las bases del conocimiento moderno en este tema28.
Durante la primera mitad del siglo xx, los embriólogos buscaban responder si el destino de una o de algunas células en particular estaba determinado genéticamente, o si el ambiente tenía alguna influencia y si éste era el caso, qué tanto; para este propósito realizaron injertos de tejido embrionario entre individuos. Un hallazgo importante fue la obtención de injertos exitosos en los cuales los individuos injertados aceptaban los tejidos extraños como propios. Llamaba la atención que cuando el receptor del trasplante era un individuo adulto, casi invariablemente ocurría el rechazo29. Estos hallazgos llevaron a concluir que los trasplantes realizados en estado embrionario eran generalmente aceptados y que esta propiedad se perdía en la edad adulta. Esta importante observación no fue debidamente valorada por los investigadores de la época ya que su interés primario era la embriología.
En 1945 R.D. Owen, quien en ese momento era investigador de la Universidad de Winsconsin, observó que gemelos dicigóticos bovinos expresaban antígenos eritrocitarios compartidos; es decir; cada gemelo generaba una importante forma de inducir tolerancia frente a los trasplantes30. Owen interpretó este hallazgo como una evidencia del intercambio genético entre células precursoras sanguíneas en el periodo embrionario. Posteriormente, Burnet y Fenner (1976) en una monografía sobre la producción de anticuerpos, plantearon que en la etapa embrionaria había un “reconocimiento de lo propio” y que si en esa etapa se introducían antígenos extraños a un individuo, éste los iba a reconocer y aceptar como propios. Simultáneamente, Peter Medawar y colaboradores trabajaban en trasplantes entre bovinos, gemelos monocigóticos y dicigóticos fraternos, y observaron que los injertos de piel entre gemelos fraternos eran exitosos, aún entre los de diferente sexo30. Este hallazgo, que no encajaba en los conceptos predominantes de la época, llevó a Medawar a retomar el trabajo de Owen y las ideas expuestas en la monografía de Burnet y Fenner enfocándose en demostrar que la tolerancia podía ser inducida. Los hallazgos obtenidos en la primera mitad de los años 50 con cepas endogámicas de ratones llevaron a la conclusión de que la tolerancia podía ser inducida en neonatos pero no en un estado adulto.
En 1956 el concepto de tolerancia inmunológica ya era ampliamente aceptado y en 1960 Burnet y Medawar recibieron el premio Nobel de Medicina por su trabajo.
Mecanismos involucrados en el desarrollo de la toleranciaHa pasado medio siglo desde que Billingham, Brent y Medawar demostraron, por primera vez, en un modelo experimental el desarrollo de la tolerancia. A partir de entonces, los inmunólogos de trasplante han buscado intensamente las causas que desencadenan la tolerancia clínica, con la esperanza de eludir las complicaciones de la inmunosupresión no específica así como los mecanismos de prevención del rechazo crónico. Los intentos por lograr entender la tolerancia clínica se basan en la comprensión de los mecanismos básicos de tolerancia. El crecimiento en el conocimiento de este concepto se ha ido desarrollando en forma paralela con la apreciación de la complejidad de los mecanismos desencadenantes de la tolerancia. En particular, se ha avanzado mucho en la comprensión del papel de las células T y B en la tolerancia central y periférica, en el papel de las células T reguladoras, así como en los mecanismos involucrados en la tolerancia de las células dendríticas31.
Tolerancia de células TTolerancia centralAl timo y a la médula ósea se les ha atribuido un papel relevante en el desarrollo de la tolerancia en las etapas embrionaria y neonatal de la vida del individuo, durante las cuales el sistema inmune desarrolla un repertorio linfocitario, que por una parte es capaz de responder de manera adecuada frente a antígenos extraños, y, por otra, en condiciones normales, tolera los antígenos propios.
El sistema inmune, en su fase inicial de maduración, identifica los linfocitos (timocitos) capaces de reconocer los antígenos propios que son presentados en el contexto de moléculas del Complejo Mayor de Histocompatibilidad (CMH), las que, a su vez, son expresadas por células retículoepiteliales y dendríticas interdigitadas ubicadas en la región corticomedular del timo. En este proceso de maduración, conocido como selección positiva, los linfocitos que reconocen con moderada afinidad los péptidos propios, logran llegar a su fase final de maduración para viajar finalmente por los conductos aferentes del timo y establecerse en los órganos linfoides secundarios. Los timocitos que no logran formar un receptor apto para el reconocimiento antigénico mueren por apoptosis32.
Entre los linfocitos que sobreviven al proceso de selección positiva hay una población que reacciona fuertemente ante los antígenos propios, la cual debe ser eliminada o modificada ya que puede ser altamente perjudicial para el individuo. Una vez identificados los linfocitos, se dan dos alternativas: eliminación por apoptosis, mecanismo conocido como selección negativa; o disminución de la afinidad del receptor, también denominado edición del receptor32,33. A continuación se explican estos dos conceptos.
•Selección negativaLa selección negativa es un proceso que se conoce con mayor detalle en el timo, en donde los autoantígenos son presentados a los linfocitos T por las células presentadoras. Hay dos factores que influyen en el destino del linfocito tímico: la afinidad del receptor por el autoantígeno, en la que a mayor afinidad mayor la probabilidad de su eliminación; y la concentración del autoantígeno en la que a mayor concentración es mayor la probabilidad de que el linfocito autorreactivo sea eliminado por selección negativa32,33.
Un descubrimiento reciente muestra que el repertorio de antígenos propios presentados en el timo es sorpresivamente grande34. Un amplio rango de genes específicos de tejido, son expresados en el timo por las células epiteliales medulares tímicas como la hormona paratiroidea, tiroglobulina, insulina y proteína C reactiva, sugiriendo que el epitelio tímico refleja lo que ocurre en la periferia35,36. Los mecanismos para esta expresión ectópica de antígenos de tejido no son bien conocidos y pueden reflejar una supresión aleatoria de trascripción génica (“modelo promiscuo”) o diferenciación epitelial en el timo (“modelo de mosaico”)37. Un hallazgo relacionado con la regulación autoinmune es el gen “autoimmune regulator”, por su sigla en inglés AIRE, implicado en el síndrome mendeliano, síndrome de poliendocrinopatía autoinmunitaria-candidiasis-distrofia ectodérmica o APECED, que ha mostrado ser activo en una subpoblación de células epiteliales tímicas medulares38.
Los defectos en la expresión de AIRE conducen al desarrollo de enfermedad autoinmune multiorgánica, dando como resultado una falla en la deleción de las células autorreactivas intratímicas39. Sin embargo, se estima que entre 102 y 103 células, expresan un antígeno en particular, el que podría limitar la capacidad de selección negativa para eliminar todos los timocitos autorreactivos, ya que es alta la probabilidad de que los timocitos reaccionen espontáneamente con antígenos ectópicos40,41. Por lo tanto, es tentador sugerir que la presentación de antígenos ectópicos en el timo puede ser un regulador importante en la generación de células T reguladoras antígeno específico, aunque actualmente no hay una clara evidencia experimental que corrobore este evento. Sin embargo, sí hay evidencias experimentales que demuestran que el timo es una fuente de células T reguladoras CD4+ específicas para los autoantígenos. Estas constituyen el 5% de la población de linfocitos maduros CD4+ CD8– y se caracterizan por presentar una alta expresión de receptores para IL-2a (IL2R) conocidos como CD25+42,43.
•Edición del receptorEl mecanismo de edición del receptor modifica la afinidad de algunos linfocitos fuertemente autorreactivos y los convierte en linfocitos T reguladores (Treg). Se postula que en este proceso están involucrados los corpúsculos de Hassall, en los que se encuentra una población de células dendríticas, que expresan una citocina similar a la IL-7 llamada linfopoyetina estromal tímica (TSLP), implicada en inducir respuestas Th2 en algunas enfermedades alérgicas. Se cree que algunos linfocitos altamente autorreactivos expresan receptores para esta citocina y que por esta vía evaden la apoptosis convirtiéndose en células reguladoras que, aunque se originan en el timo, actúan en los órganos periféricos. Por tal razón, las células reguladoras hacen parte del mecanismo de supresión, que a su vez hace parte de la tolerancia periférica44.
Tolerancia periféricaAunque la tolerancia central es bastante eficaz, es posible observar linfocitos fuertemente autorreactivos que han escapado a ella. Se han identificado mecanismos adicionales de control que actúan en los órganos periféricos. Estos mecanismos se han clasificado bajo el nombre de tolerancia periférica. A diferencia de la tolerancia central, los mecanismos de tolerancia periférica actúan durante toda la vida del individuo y lo hacen sobre poblaciones de linfocitos maduros completamente desarrollados. Dentro de los mecanismos involucrados en la tolerancia periférica se encuentran: la anergia, la inmunoignorancia, la deleción clonal periférica y las células T reguladoras44. Veamos:
•AnergiaPara entender el mecanismo de la anergia es necesario recordar que la activación del linfocito T requiere de varias señales: la primera, en la que el antígeno es presentado al receptor de la célula T por la célula presentadora de antígeno, en el contexto de la molécula del complejo mayor de histocompatibilidad; y la segunda, por moléculas coestimuladoras, ej: B7.1 y B7.2 en la célula presentadora de antígeno, las cuales son reconocidas por moléculas CD28 en el linfocito T. Cuando se presenta un autoantígeno en condiciones normales, sólo se da la señal 1 y sin coestimulación o señal 2, lo que lleva a una falta de respuesta del linfocito T. Por el contrario, cuando es un antígeno extraño el que es presentado, generalmente la respuesta se da en un contexto inflamatorio o infeccioso donde las señales coestimulatorias son abundantes, dando como resultado la activación del linfocito T44,45.
Otra molécula implicada en este mecanismo es la proteína Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), que compite con CD28 por la unión con moléculas contraparte CD80(B7-1) y CD86 (B7-2), ubicadas en la superficie de las células presentadoras (CPA). Cuando la expresión de CD28 es baja, predomina la expresión de CTLA-4 y el resultado es la inactivación del linfocito T. Otro receptor inhibitorio importante es el PD-1 cuya mutación está asociada en modelos murinos con enfermedades autoinmunes como la artritis44.
Una forma de inducción de anergia es mediante la utilización de moléculas bloqueadoras de los coestimuladores. Un ejemplo de esto son los anticuerpos anti CD28 y anti CD154 que han sido utilizados para mejorar la supervivencia de trasplantes30.
•IgnoranciaSe caracteriza por la ausencia de activación linfocitaria a pesar de la existencia de linfocitos autorreactivos. Han sido planteadas dos posibles explicaciones a este fenómeno: la primera es que el autoantígeno está en muy bajas concentraciones, insuficientes para emitir una adecuada primera señal; la segunda tiene que ver con la existencia de sitios inmunológicamente privilegiados como el sistema nervioso central, la cámara anterior del ojo, los testículos, que no permiten un amplio contacto entre los antígenos presentes en ellos con el sistema inmune28.
•Deleción clonal periféricaEste mecanismo implica la muerte de los linfocitos autorreactivos al hacer contacto con el antígeno propio con una alta especificidad. La molécula Fas, perteneciente a la familia del receptor del factor de necrosis tumoral (TNF), ha sido identificada como un receptor de muerte celular en linfocitos, que activa la apoptosis al encuentro con FasL, un homólogo de TNFa. A continuación se activa el gen Bim, que es un miembro proapoptótico de la familia Bcl-2 el cual desencadena la activación de las caspasas, que llevan a la muerte de la célula46. Este mecanismo se ha aplicado en la inmunología de trasplantes al inducir la expresión de FasL en alotrasplantes de islotes pancreáticos a ratones diabéticos logrando una mejor supervivencia del trasplante27.
Otra estrategia, utilizada en monos, ha sido la de ligar una antitoxina a un anticuerpo monoclonal dirigido contra CD3; de este modo se ha logrado prolongar la supervivencia de trasplantes de riñón y piel en los animales mencionados47.
•Células T reguladorasLos mecanismos de tolerancia periférica, mencionados hasta el momento, se han llamado mecanismos “recesivos” debido a que en todos ellos hay una regulación intrínseca de la célula con potencial autorreactivo. El mecanismo de regulación dominante, anteriormente llamado supresión, por el contrario, proviene de otra célula44,48.
Las primeras evidencias de la existencia de células reguladoras provienen de modelos animales con autoinmunidad. Estos modelos podían ser espontáneos (animales con una susceptibilidad predeterminada) o inducidos (por ejemplo, la extirpación del timo en ratones neonatos que genera la aparición de diversas enfermedades autoinmunes). Este tipo de experimentos llevó a sugerir que el sistema inmune tenía el potencial de desarrollar procesos autoinmunes, pero también que habían componentes celulares que eran capaces de controlar dichos procesos. Inicialmente, se identificó una población linfocitaria con baja expresión de CD45, CD25, y alta expresión de CD5 asociadas con la aparición de enfermedades autoinmunes en modelos murinos, observándose un retorno a la normalidad cuando se restablecía la población con células con fenotipos normales.
Posteriormente, Sakaguchi et al. (2008), identificaron poblaciones celulares de linfocitos T carentes del marcador CD25 implicadas en la aparición de autoinmunidad en ratones atímicos, en tanto que el aporte de linfocitos CD25+ claramente revertía el proceso44,45. Al estudiar esta población celular, se observó la presencia de un factor de transcripción conocido como FOXP3, producto de la expresión del gen FOXP3 ubicado en el cromosoma X en pacientes con enfermedad denominada síndrome de mala regulación inmune, poliendocrinopatía y enteropatía ligada al X (IPEX) la cual se manifestaba en niños varones con mutación en el gen FOXP349.
Actualmente, no hay consenso de cómo las células reguladoras CD4+ CD25+ maduran en el timo y de la forma como los péptidos propios realizan la selección de estas células. Las células epiteliales corticales tímicas parecen ser necesarias para la generación de este repertorio49. La generación de timocitos reguladores FOXP3 requiere de la afinidad del TCR por los péptidos propios, y de la presencia de IL-2; su ausencia no puede ser reemplazada por otras citocinas. Sin embargo, FOXP3 regula negativamente la producción de IL-2, IL-4 y positivamente la producción de CTLA-4 y CD25, a través de su interacción con factores de transcripción como NFAT y NF-B, entre otros44,45.
Aunque los trabajos iniciales fueron desarrollados en modelos murinos, varios grupos han identificado células reguladoras en humanos CD4+ CD25+50–52.
Las células T reguladoras pueblan los órganos linfoides secundarios, los tejidos periféricos y los sitios de inflamación, en donde disminuyen la activación de las células T, células NK, células dendríticas y células B, a través de mecanismos poco conocidos e induciendo tolerancia inmunológica53,54. Una notable disminución en las poblaciones de T reguladoras y defectos funcionales, se han reportado en un gran número de enfermedades autoinmunes incluyendo lupus eritematoso sistémico, diabetes tipo 1 y artritis reumatoide55.
Durante los últimos años se han identificado varias subpoblaciones de linfocitos T reguladores, entre las que se encuentran:
- •
Células T reguladoras CD4+ CD25+, FOXP3+, conocidas como naturales.
- •
Células T inducidas FOXP3 (–), conocidas como Tr1 que son activadas por interleucina 10 (IL10).
- •
Células Th3 que expresan TGF-b. Estas poblaciones se caracterizan por tener un fenotipo FOXP3 (–); sin embargo, algunas de ellas expresan FOXP3.
- •
- •
Células reguladoras iNKT, llamadas así por expresar marcadores de membrana, tanto de células T como de células NK. El receptor TCR de estas células presenta mucho menos variabilidad que el TCR de las células T comunes. Las células iNKT son capaces de aumentar la secreción de citocinas reguladoras, especialmente IL-4 y por esta vía ser capaces de limitar la respuesta inmune56,57.
- •
Se plantea la existencia de células presentadoras de antígeno con actividad reguladora58.
Los mecanismos por medio de los cuales las células T reguladoras controlan la actividad de las células B, no son muy claros. Sin embargo, algunos investigadores han reportado que las células T reguladoras pueden inducir tolerancia a respuestas de tipo adaptativo mediante la inducción de la muerte de las células B, la supresión en la producción de anticuerpos y la regulación de la supervivencia de células plasmáticas57,59–61. En modelos murinos con lupus eritematoso sistémico inducido (caracterizado por una excesiva producción de anticuerpos), se ha demostrado que un aumento en la población de T reguladoras suprime la enfermedad mediante la limitación en la producción de autoanticuerpos62,63.
Tolerancia de células BAl igual que en las células T, el desarrollo de las células B se genera a partir de un repertorio linfocitario responsable de la diversidad de la respuesta inmune; sin embargo, dentro de este repertorio también aparecen células B autorreactivas que, de no ser controladas, pueden ocasionar respuestas perjudiciales para el individuo.
Los mecanismos de tolerancia en células B pueden ser divididos en dos: primarios o de novo y mecanismos secundarios64. Los mecanismos primarios se caracterizan porque se presentan durante la diferenciación de las células B y contribuyen a formar el repertorio linfocitario; en tanto que los mecanismos secundarios no alteran dicho repertorio, sino que modulan la respuesta de los linfocitos B maduros en los órganos linfoides periféricos.
Mecanismos primarios (de novo)Dependen de una combinación entre la avidez antigénica y el umbral de activación del receptor de célula B (BCR). Ocurren tanto en los órganos primarios (médula ósea, hígado fetal) como en los tejidos linfoides periféricos. En los primeros se denominan mecanismos de tolerancia central y en los segundos mecanismos de tolerancia periférica.
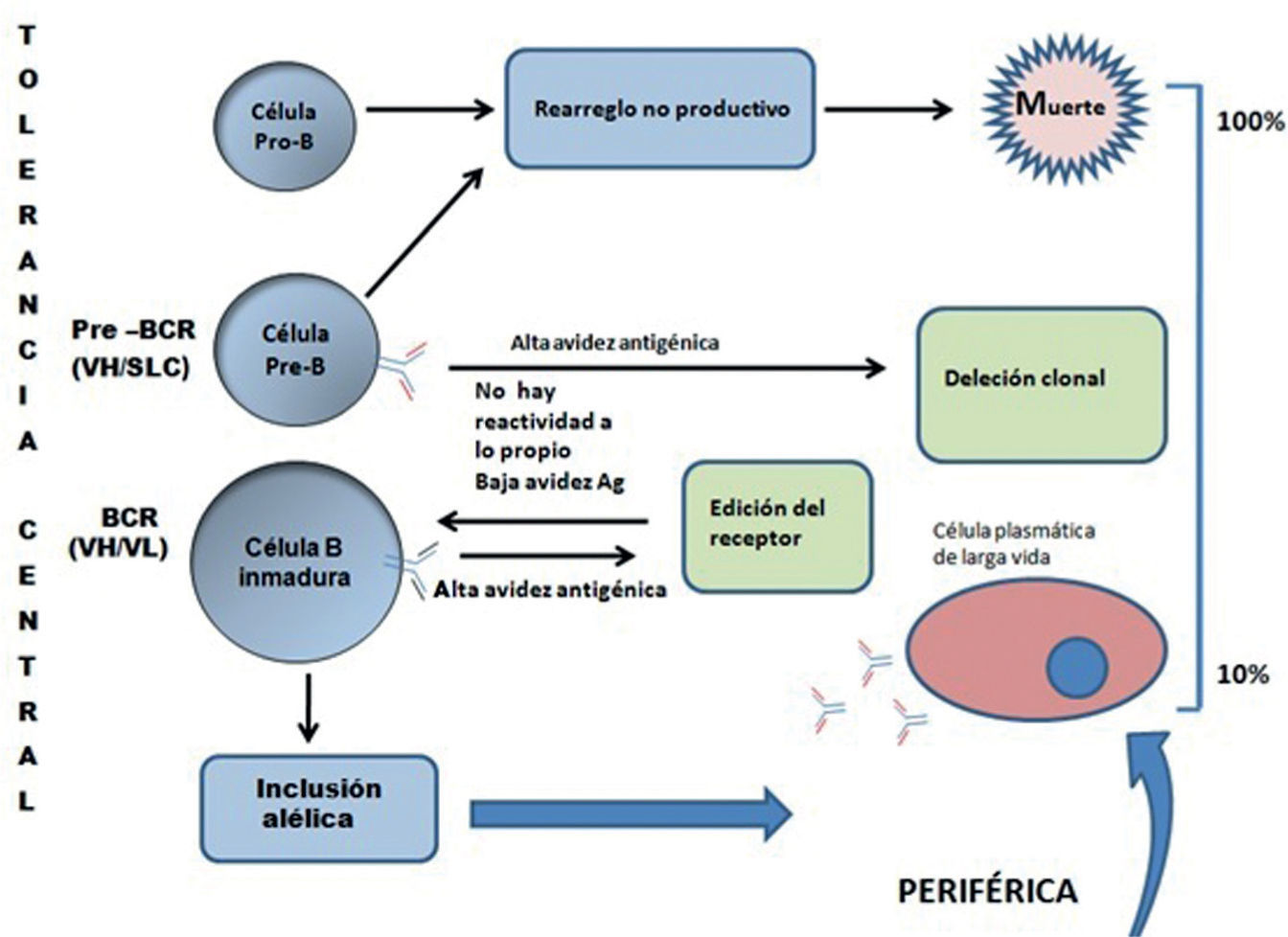
Tolerancia centralEl desarrollo de los linfocitos B ocurre en sus etapas iniciales en la médula ósea y pasa por diferentes estadios de maduración: Pro B, Pre B y B inmaduro. Estos estadios cursan con cambios en la expresión de receptores de membrana dentro de los cuales llama la atención la expresión del receptor de la célula B (BCR), que no se expresa en estadios iniciales. En el estadio Pro B, BCR, se expresa de manera provisional. En el estadio de célula Pre B se expresa con la cadena liviana sustituta invariable y aparece en su forma final en la célula B inmadura (fig. 1).
 con cadenas ligeras sustitutas (SLC) son delecionadas. BCR: receptor de células B.")
Maduración de los linfocitos B en la médula ósea. Etapas de maduración: célula Pro B a Pre B, y linfocito B inmaduro, en donde se aprecian los cambios durante la ontogenia de la célula, relacionados con la avidez y maduración central y periférica. Las células Pre B que expresan cadenas pesadas de inmunoglobulinas altamente autorreactivas (VH) con cadenas ligeras sustitutas (SLC) son delecionadas. BCR: receptor de células B.
En el paso de linfocito Pre B a linfocito B inmaduro, ocurre la recombinación somática del segmento variable de la cadena liviana de la inmunoglobulina, que va a reemplazar la cadena liviana sustituta invariable antes descrita. En este punto, con el BCR completo, los linfocitos con alta avidez por autoantígenos son eliminados por deleción clonal central y los que presentan moderada avidez por autoantígenos experimentan el mecanismo conocido como edición del receptor, en el que los genes RAG, responsables de la recombinación somática, se reactivan momentáneamente y disminuyen la avidez del receptor por autoantígenos pero conservando un receptor funcional frente a antígenos extraños. Hoy en día, se considera que la edición del receptor es el mecanismo más importante utilizado en la tolerancia central, por encima de la deleción clonal central65.
Otro fenómeno recientemente descrito es el conocido como “inclusión alélica”, que se refiere a que en este punto del desarrollo de los linfocitos B, el 50% de ellos expresa 2 o más cadenas livianas diferentes, en contraposición al concepto de “exclusión alélica” que postula que un linfocito maduro expresa sólo una forma de cadena liviana y una forma de cadena pesada en su receptor, esto es, que las dos cadenas livianas que conforman el BCR, son iguales. Lo mismo es cierto para las cadenas pesadas.”La inclusión alélica“explica porqué un número importante de linfocitos autorreactivos escapan a la edición del receptor y se encuentran en los órganos periféricos66.
Llama la atención que en el mecanismo de edición del receptor podrían tener que ver, además de la alta avidez del BCR por el autoantígeno, otros receptores del sistema inmune innato, como el de la IL-1 y los Toll Like Receptor (TLR)67. Esto se plantea porque pacientes con defectos en moléculas que tienen que ver con las vías de señalización de los receptores antes mencionados (como IRAK-4, MyD88 y UNC 93B), presentan fallas en los mecanismos de tolerancia central y periférica, y se observa en ellos mayor cantidad de linfocitos B autorreactivos.
No se ha observado en la médula ósea un equivalente al gen AIRE que medie la expresión en el timo de antígenos que se expresan de forma restringida en ciertos órganos (por ejemplo el páncreas). Sin embargo, se piensa que una expresión de este tipo de antígenos ocurre en la médula ósea, lo que permite que la mayoría de linfocitos B sean eliminados en este sitio68.
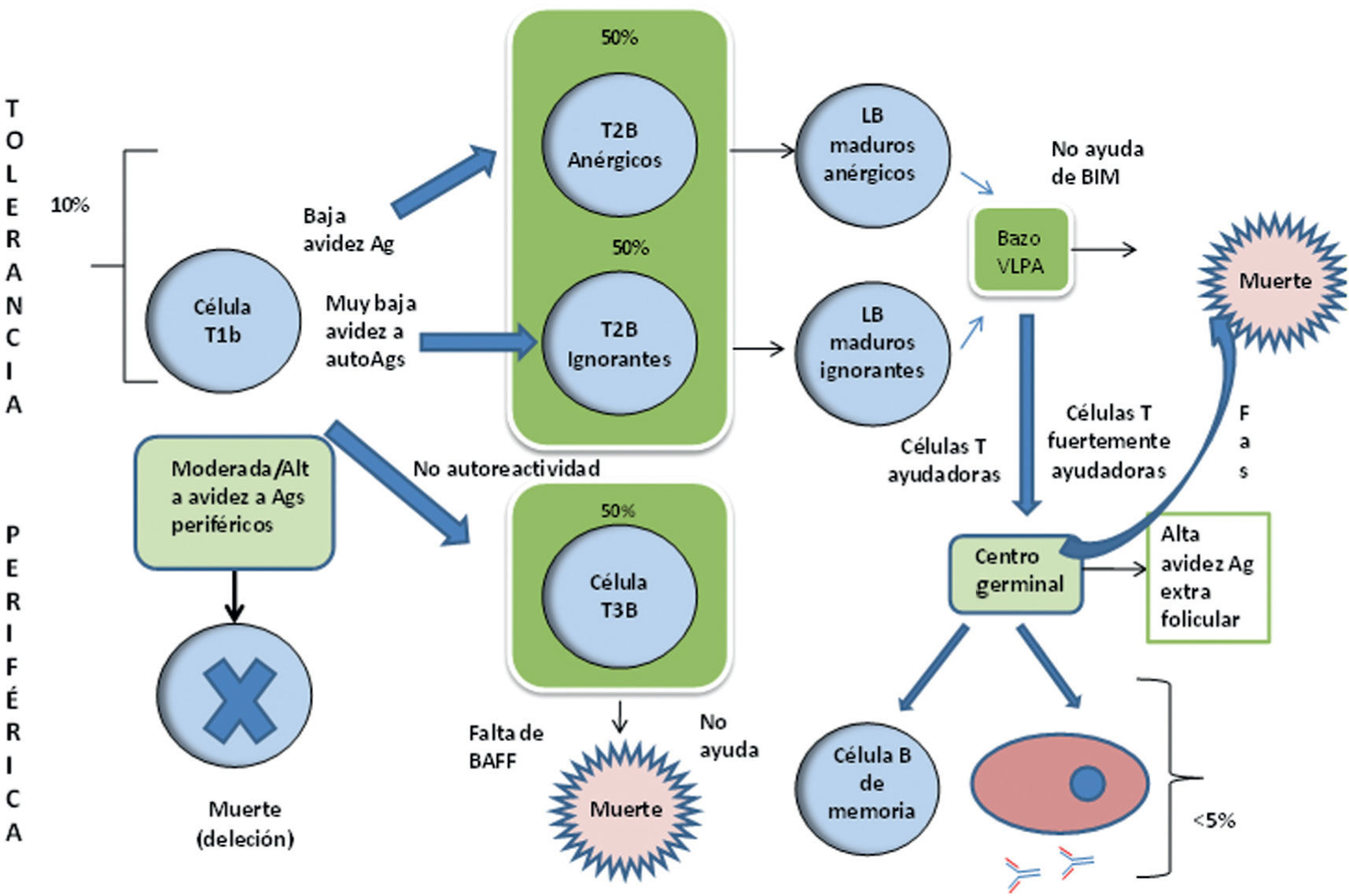
Tolerancia periféricaEl 10% de linfocitos generados en la médula ósea sobreviven a los mecanismos de tolerancia central y migran al bazo como linfocitos transicionales o linfocitos T1B. Allí los linfocitos T1B se encuentran con autoantígenos que no estaban presentes en la médula ósea. Los linfocitos T1B que presentan avidez moderada o alta por estos autoantígenos, son eliminados por una vía dependiente de Bim, en tanto que los linfocitos T1B con una avidez baja o muy baja por los autoantígenos, entran en anergia o ignorancia. Bajo una exposición posterior al autoantígeno o antígeno extraño, estas células ahora conocidas como T2B se dirigen a las vainas linfoides periarteriolares (VLPA), que son las zonas de predominio de células T en el bazo, donde si no reciben suficiente estimulación por parte de las células T o señales coestimulatorias dependientes de TLR, mueren en 2 o 3 días (fig. 2).
, que son las zonas de predominio de células T en el bazo, donde si no reciben suficiente estimulación por parte de las células T o señales coestimulatorias dependientes de TLR, mueren en 2 o 3 días. Las células T1b que se diferencian en T3B pueden entrar en apoptosis ante la falta de BAFF. BAFF: factor activador de células B; BIM: miembro de la familia BCL2 inductor de apoptosis.")
Estadios de maduración de linfocitos B en médula ósea y bazo. El 10% de linfocitos generados en la medula ósea que sobreviven a los mecanismos de tolerancia central migran al bazo como linfocitos transicionales o linfocitos T1B. Los linfocitos T1B se encuentran con autoantígenos que no están presentes en la medula ósea. Los linfocitos T1B que presentan avidez moderada o alta por estos autoantígenos, son eliminados por la vía dependiente de BIM, en tanto que los linfocitos T1B con avidez baja o muy baja por autoantígenos entran en anergia o ignorancia. Ante una exposición posterior al autoantígeno o antígeno extraño, estas células ahora conocidas como T2B se dirigen a las vainas linfoides periarteriolares (VLPA), que son las zonas de predominio de células T en el bazo, donde si no reciben suficiente estimulación por parte de las células T o señales coestimulatorias dependientes de TLR, mueren en 2 o 3 días. Las células T1b que se diferencian en T3B pueden entrar en apoptosis ante la falta de BAFF.
BAFF: factor activador de células B; BIM: miembro de la familia BCL2 inductor de apoptosis.
Las células T2B anérgicas pueden revertir esta condición si son expuestas a fuertes dosis del factor de supervivencia de células B (BAFF), dosis altas de antígeno o estimulación fuerte de células T. Si lo anterior ocurre, estas células anérgicas sobreviven y se dirigen a los centros germinales (CG) junto con las células B ignorantes69. La falla en el mecanismo de anergia ha sido implicada en la aparición de enfermedades autoinmunes como el lupus eritematoso sistémico y el síndrome de Sjögren70.
Mecanismos secundariosLos mecanismos secundarios se dividen en intrínsecos y extrínsecos al sistema inmune. Dentro de los primeros están las citoquinas, anticuerpos, células reguladoras (B y T), macrófagos, sistema de complemento y vías de señalización ligadas al BCR. Mientras que los segundos tienen que ver con agentes externos (derivados de patógenos) o con células propias mutadas como es el caso de los tumores.
Los mecanismos de tolerancia pueden ser afectados por factores relacionados con el antígeno o con el huésped. Los últimos trabajos experimentales han reportado defectos genéticos que llevan a fallas en la tolerancia central y periférica71. Por otra parte, la suma de diversos defectos, que por separado no serían suficientes para generar problemas de autoinmunidad, termina ocasionando este tipo de problemas cuando confluyen al tiempo en un mismo paciente72. En estudios recientes se ha demostrado la existencia de células B reguladoras (B regs) en ratones y se postula su existencia en humanos. Las células B regs no tienen un marcador homólogo al FOXP3 que se encuentra en las células T reguladoras, pero presentan alta expresión de CD1d, CD21, CD24 e IgM y una moderada expresión de CD19. Por otra parte, los linfocitos B regs producen IL-10, al igual que algunos linfocitos T reguladores. La caracterización de estas células en humanos puede traer nuevas aproximaciones terapéuticas en el campo de la tolerancia a trasplantes73.
Tolerancia mediada por células dendríticasLas células dendríticas (CDs) constituyen una población heterogénea de células profesionales presentadoras de antígenos. Subpoblaciones reguladoras de CDs han sido identificadas en el bazo y nódulos linfoides de murinos que incluyen: CDs CD11c+, CDs CD8α+, CDs CD4+ CD8– y CDs CD4-CD8–74, mientras que en humanos las subpoblaciones de CDs incluyen CDs mieloides (mCDs) y plasmocitoides (pCDs)75–77. En los últimos años, un homólogo murino de pCDs ha sido identificado, el que no expresa marcadores de células T ni de B ni marcadores mieloides, y exhibe cierta similitud morfológica con células plasmáticas78.
Las CDs tienen la capacidad de inducir inmunidad o tolerancia dependiendo de su estado de activación, de señales intracelulares y de las citoquinas que estén en el entorno79,80. Debido a su doble funcionalidad (inducción de la inmunidad o tolerancia), han sido el blanco de terapias inmunológicas para el tratamiento de tumores e inducción de tolerancia en enfermedades autoinmunes y trasplantes. Las CD inmaduras (imCD) no pueden generar señales coestimuladoras durante la activación de los linfocitos T, lo que resulta en un mecanismo de inducción de tolerancia de las células T81. La enzima inmunorreguladora indoleamina 2,3-dioxigenasa (IDO), que cataliza la degradación del triptófano, contribuye a la tolerancia inmunológica82–88. La IDO-expresada por las CDs es capaz de suprimir la proliferación de células T y promover la apoptosis de las células89–93.
Varias subpoblaciones de CDs se han relacionado con el desarrollo de células T reguladoras. Las CD intratímicas se han visto implicadas en el desarrollo de las células T reguladoras naturales94. In vitro, las CD derivadas de imCD, inducen la diferenciación de células T reguladoras FOXP3 +95 y Tr196. Recientemente, una población especializada de CD con fenotipo CD103+ aislada de ganglios linfáticos mesentéricos y de la lámina propia de ratones normales, se ha visto involucrada en la conversión de células T naive en células T reguladoras FOXP3+97,98. Este proceso de conversión es dependiente de TGFb y de ácido retinoico99.
Las terapias basadas en la obtención de CDs tolerogénicas pueden ser desarrolladas in vitro utilizando IL-10, TGFb, prostaglandina E2, vitamina D3 o rapamicina, las CDs tolerogénicas generadas in vitro pueden mediar la inmunosupresión e inducir tolerancia inmune, a través de múltiples mecanismos, como la baja regulación en la expresión de moléculas coestimuladoras, alteración de la polarización hacia células T helper y la expansión de células T reguladoras a partir de células T naive100–103.
Dado que las células dendríticas juegan un papel central en la inducción, tanto de la inmunidad como en la tolerancia, es posible que la modulación de la plasticidad de las células dendríticas pueda ser utilizada para la inducción de tolerancia en trasplantes y en enfermedades autoinmunes. Varios grupos de investigación han desarrollado diversos métodos que permiten manipular y mantener células dendríticas in vitro. La actividad de las CDs se puede modular de las siguientes maneras: en presencia de citocinas (GM-CSF, IL-10, o TGFb)99; utilizando técnicas de ingeniería genética; induciendo la expresión transgénica de IL-10; TGFb, o IDO103–106; silenciando algunos genes como RelB; induciendo la expresión de genes para IL-1295,96; utilizando mediadores farmacológicos; empleando drogas antiinflamatorias como rapamicina y ciclosporina; administrando vitamina D3, prostaglandina E2 o ligandos para el receptor inhibitorio de la familia de las inmunoglobulinas ILT4107,108.
El uso de citoquinas como G-CSF, GM-CSF o Flt 3-ligando, involucradas en la expansión de CDs tolerogénicas y otras células mieloides, ha generado cierto nivel de éxito debido a que pueden prevenir el rechazo de aloinjertos. Sin embargo, la eficacia varía debido a la generación de múltiples tipos de células dentro de los cultivos celulares, a la dosis de citoquinas o la duración de la estimulación con citoquinas109. Otros grupos han utilizado rapamicina o compuestos que inhiben la maduración de CDs inm a CDs tolerogénicas110.
ConclusionesDesde los trabajos de Brent y Medawar hasta nuestros días, el estudio de los mecanismos involucrados en la tolerancia inmunológica a trasplantes y antígenos de microorganismos sigue siendo un tema central de los inmunólogos. Para algunos, la pérdida de la tolerancia sería responsable del desarrollo de las enfermedades autoinmunes, lo que a su vez ejemplifica la dicotomía del sistema inmune. A la suma de eventos generados en el timo y la médula ósea durante el desarrollo de células T y B, se le atribuye un papel relevante en la tolerancia inmunológica. Por otra parte, los mecanismos de tolerancia que ocurren en los órganos periféricos, complementan los de tolerancia central y evitan, en condiciones normales, la aparición de las enfermedades autoinmunes. En los últimos años, diferentes investigaciones en poblaciones celulares, como las células epiteliales tímicas, las células dendríticas tolerogénicas y las poblaciones de linfocitos T reguladores, han arrojado nuevas evidencias que han ayudado a entender, de una mejor manera, esta área fundamental de la inmunología; sin embargo, hay aún mucho camino por recorrer para esclarecer las verdaderas causas del desarrollo de la tolerancia, con el fin de generar estrategias terapéuticas tanto en el control del rechazo al trasplante como en la prevención o el tratamiento de la enfermedad autoinmune.
Los autores declaran no tener ningún conflicto de intereses.
Agradecimientos
A Jose Julián y Juan Sebastián Siachoque Jara, estudiantes de la facultad de Medicina de la Universidad del Rosario por sus aportes en la elaboración del manuscrito.
