



La dermatomiositis es una miopatía inflamatoria idiopática que se caracteriza por presentar lesiones en la piel; por su presentación clínica, su curso y su pronóstico, se la considera una enfermedad heterogénea. En Colombia existen pocos registros que describan las características clínicas de los pacientes afectados por esta enfermedad.
MétodosEstudio descriptivo de corte transversal, se revisaron las historias clínicas de pacientes que consultaron a un hospital universitario en Colombia entre enero del 2004 y diciembre del 2019. Los registros se obtuvieron utilizando bases de datos de las unidades de dermatología, reumatología, dermatopatología, electrofisiología y códigos diagnósticos CIE10 asociados con dermatomiositis.
ResultadosSe obtuvieron 70 pacientes con diagnóstico de dermatomiositis, 63 (90%) de los cuales cumplían criterios de clasificación de Bohan y Peter, en tanto que 7 (10%) presentaban dermatomiositis amiopática. El promedio de edad fue de 43 años (DS±15,3); 48 fueron mujeres (68,5%); los signos clínicos más frecuentes fueron: pápulas de Gottron (80%, n=56), eritema heliotropo (78,5%, n=55) y poiquilodermia (75,7%, n=53). Las manifestaciones sistémicas más comúnmente encontradas fueron: disfagia (21,4%, n=15), enfermedad pulmonar intersticial (11,4%, n=8) e hipertensión pulmonar (8,5%, n=6). Se documentó cáncer en el 8,5% (n=6) de los pacientes.
ConclusiónSe presenta información clínica de pacientes con dermatomiositis en un centro hospitalario de referencia en Colombia; los datos obtenidos concuerdan con la información de otros estudios de series de casos a escala mundial.
Dermatomyositis is an idiopathic inflammatory myopathy characterized by the presence of skin lesions; it is considered a heterogeneous disease, due to its clinical presentation, course, and prognosis. In Colombia there are few records that describe the clinical characteristics of these patients.
MethodsCross-sectional study. Medical records of patients who consulted a university hospital in Colombia between January 2004 and December 2019 were reviewed. The records were obtained using databases from the dermatology, rheumatology, dermatopathology, and electrophysiology units, and CIE10 diagnostic codes.
ResultsSeventy patients with a dermatomyositis diagnosis were found, 63 (90%) fulfilled the Bohan and Peter diagnostic criteria and 7 (10%) had amyopathic dermatomyositis, with an average age of 43 years (SD±15.3). Forty-eight were women (68.5%). The most frequent clinical signs were Gottron's papules 80%, periorbital violaceous (heliotrope) erythema with oedema 78.5% (n=55) and poikiloderma 75.7% (n=53). The most frequently found systemic manifestations were dysphagia (21.4%, n=15), interstitial lung disease (11.4%, n=8), and pulmonary hypertension (8.5%, n=6). Cancer was documented in 8.5% (n=6) of patients.
ConclusionWe showed clinical information of patients with dermatomyositis in a referral hospital in Colombia. The data obtained is consistent with information from other case series worldwide.
Las miopatías inflamatorias son consideradas enfermedades raras, sin embargo, dentro de ellas, la dermatomiositis es la más común, excepto en pacientes de edad avanzada, en quienes predomina la miositis por cuerpos de inclusión1. La dermatomiositis es la única miopatía inflamatoria que se caracteriza por presentar lesiones en la piel, y puesto que las manifestaciones dermatológicas pueden constituir los primeros hallazgos de la enfermedad, deben ser reconocidas clara y oportunamente2. En los EE. UU., la prevalencia estimada es de 1-6 pacientes por 100.000 habitantes3. En Colombia, la incidencia y la prevalencia se desconocen.
La dermatomiositis se considera una enfermedad heterogénea, no solo por su presentación clínica, sino también por su curso y pronóstico4. Además del músculo y la piel, puede comprometer diferentes órganos y sistemas, como el corazón, el tracto gastrointestinal y los pulmones, lo que la convierte en un reto diagnóstico5. Se puede asociar con malignidad, y se puede presentar de manera concomitante con otras enfermedades del tejido conectivo4.
A pesar de que se clasifica dentro de las enfermedades raras, tiene una importante morbimortalidad, que puede explicarse por su evolución crónica y progresiva, con un daño muscular irreversible. Hasta el momento, existen escasos datos que permitan caracterizar a los pacientes que sufren de dermatomiositis en Colombia, y en vista de la importancia de un diagnóstico y un manejo oportunos, este trabajo busca describir las principales características demográficas, las manifestaciones dermatológicas y sistémicas, el perfil inmunológico y la concomitancia con malignidad y otras enfermedades de tejido conectivo, en un hospital universitario en Bogotá, Colombia.
MétodosEstudio descriptivo de corte transversal que se llevó a cabo en un hospital universitario en Bogotá, Colombia. El estudio incluyó pacientes con diagnóstico de dermatomiositis que cumplían los criterios de clasificación de Bohan y Peter, durante el periodo comprendido entre enero del 2004 y diciembre del 2019.
Los datos se obtuvieron a partir de diferentes fuentes de información, dentro de las que se encuentra el sistema de historias clínicas electrónicas del hospital, en el que se buscaron códigos diagnósticos CIE10 asociados a dermatomiositis (M33: Dermatopolimiositis, M330: Dermatomiositis juvenil, M331: Otras dermatomiositis, M332: Polimiositis y M339: Dermatopolimiositis, no especificada). Además, se utilizaron las bases de datos de las unidades de dermatología, reumatología, dermatopatología y electrofisiología de nuestro hospital.
Se evaluaron variables clínicas y paraclínicas, empleando medidas de resumen en frecuencias y porcentaje para variables cualitativas, así como medidas de tendencia central con promedios y dispersión para variables cuantitativas, utilizando en este caso Microsoft Excel®.
Consideraciones éticasEl estudio fue aprobado por el Comité de Ética e Investigación de la Facultad de Medicina de la Pontificia Universidad Javeriana, con número de aprobación 2019/207. El comité de ética consideró que debido a que se trataba de un estudio retrospectivo, sin intervención específica ni riesgos para los pacientes, no requería la firma de un consentimiento informado.
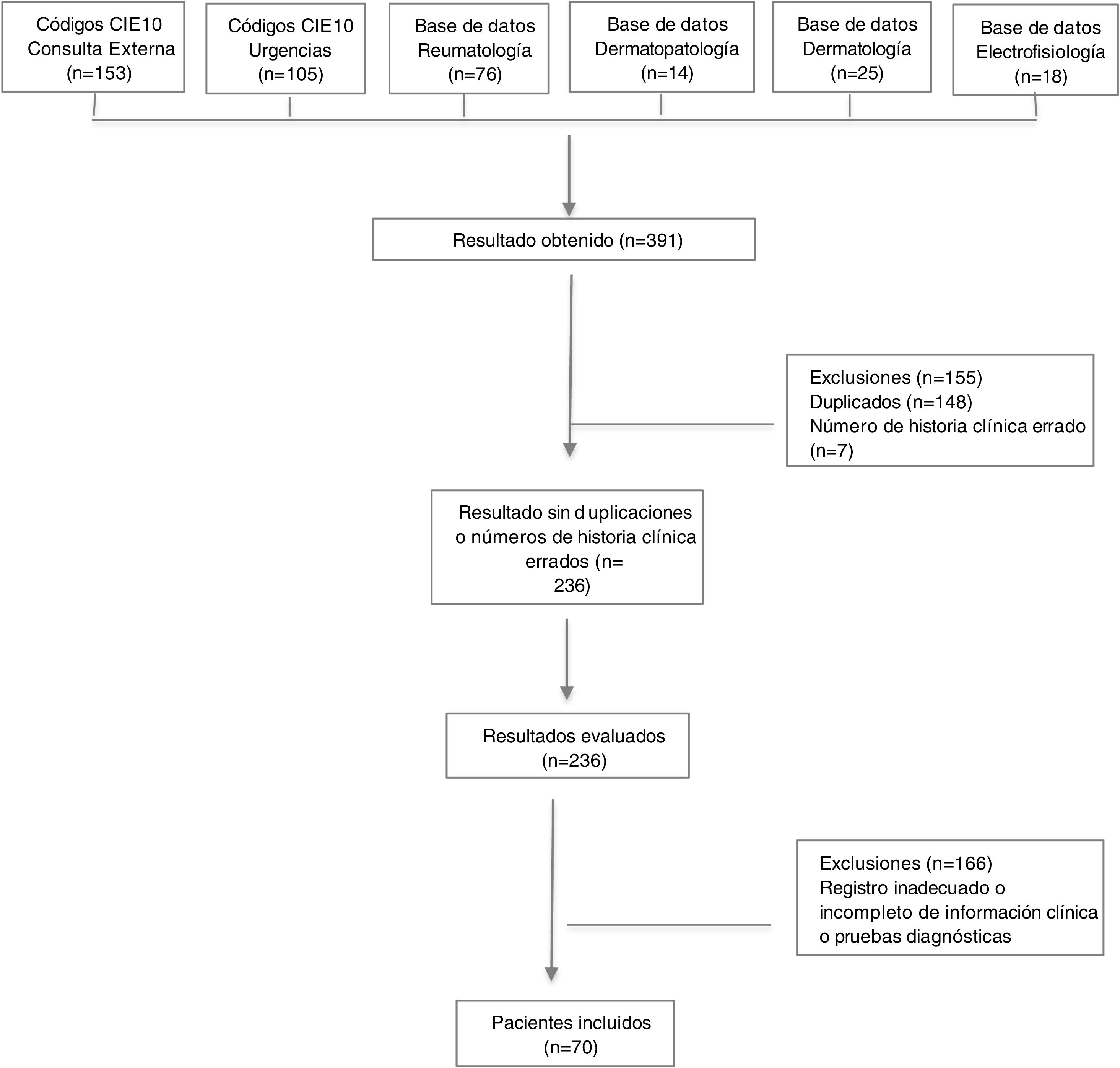
ResultadosAspectos demográficos generalesSe obtuvieron 236 resultados, de los cuales 63 pacientes cumplían los criterios de clasificación de Bohan y Peter, y siete presentaban diagnóstico de dermatomiositis amiopática. Se excluyeron números de historias clínicas duplicados y errados, además de pacientes con diagnóstico intrainstitucional de dermatomiositis, pero con registro inadecuado o incompleto de la información relacionada con las variables de interés, así como con los pacientes con diagnóstico extrainstitucional que no contaban con reporte en la historia clínica de pruebas diagnósticas que permitieran confirmar el diagnóstico (fig. 1).

El promedio de edad de diagnóstico fue de 43 años (DS±15,3), 48 fueron mujeres (68,5%), con una relación mujer:varón 2,1:1. Del total de pacientes, 63 (90%) presentaban dermatomiositis y 7 presentaban dermatomiositis amiopática (10%). Asimismo, 59 (84,2%) pacientes requirieron al menos una hospitalización; en promedio, 1,9 hospitalizaciones (±2,4), con estancia hospitalaria de 8,6 días (±10,1) (tabla 1).
Características demográficas
| Clásica | Amiopática | Total | |
|---|---|---|---|
| Número de pacientes, n (%) | 63 (90) | 7 (10) | 70 |
| Edad de diagnóstico, años (DE) | 43 (± 15,3) | 47 (± 7,4) | 43 (± 15,3) |
| Dermatomiositis en adultos, n (%)Dermatomiositis juvenil, n (%) | 60 (85,7)3 (4,2) | 7 (10) | 67 (95,7)3 (4,2) |
| Género | |||
| Femenino, n (%)Masculino, n (%) | 43 (61,4)20 (28,5) | 7,12,8 | 48 (68,5)22 (31,4) |
| Hospitalizaciones, n (%) | 2,0 (± 2,5) | 0,8 (± 0,3) | 1,9 (± 2,4), |
| Estancia hospitalaria, días (DE) | 8,6 (± 10,3) | 8,4 (± 8,9) | 8,6 (± 10,1) |
| Mortalidad, n (%) | 5 (7,14) | - | 5 (7,14) |
DE: desviación estándar.
En cuanto a los hallazgos dermatológicos encontrados en los pacientes con dermatomiositis clásica, 63 (100%) pacientes presentaron manifestaciones patognomónicas1,6, dentro de las cuales las pápulas de Gottron y el eritema en heliotropo fueron las más frecuentes, documentándose ambos hallazgos en 49 (77,7%) pacientes.
Cuarenta (63,4%) pacientes presentaron manifestaciones características. El signo de la V, que fue la más frecuente, se encontró en 30 (47,6%) de ellos. Las manifestaciones compatibles se documentaron en 51 (80,9%) pacientes, mientras que 7 casos (11,1%) correspondieron a las manifestaciones infrecuentes, 10 (15,8%) a las raras y 23 (36,5%) a las inespecíficas (tabla 2).
Manifestaciones dermatológicas
| Clásica | Amiopática | |
|---|---|---|
| Manifestaciones dermatológicas patognomónicas, n (%) | ||
| Pápulas de Gottron | 49 (77,7) | 7 (100) |
| Signo de Gottron | 25 (39,6) | 4 (57,1) |
| Eritema en heliotropo | 49 (77,7) | 6 (85,7) |
| Manifestaciones dermatológicas características, n (%) | ||
| Telangiectasias periungueales | 17 (26,9) | 3 (42,8) |
| Hipertrofia periungular | 16 (25,3) | 2 (28,5) |
| Infartos hemorrágicos en los pliegues | 1 (1,5) | 1 (14,2) |
| Signo del chal | 19 (30,1) | 2 (28,5) |
| Signo de la V | 30 (47,6) | 3 (42,8) |
| Placas atróficas, descamativas y eritematosas en el cuero cabelludo | 3 (4,7) | — |
| Signo de la funda de pistola | 11 (17,4) | 1 (14,2) |
| Manifestaciones dermatológicas compatibles, n (%) | ||
| Poiquilodermia | 47 (74,6) | 6 (85,7) |
| Edema periorbital y facial | 19 (30,15) | 1 (14,2) |
| Manifestaciones dermatológicas infrecuentes, n (%) | ||
| Vésico-ampollas epidérmicas | 1 (1,5) | — |
| Lesiones necróticas o erosiones superficiales | 3 (4,7) | — |
| Vasculitis cutánea | 3 (4,7) | — |
| Calcinosis cutis | 3 (4,7) | 1 (14,2) |
| Manifestaciones dermatológicas raras, n (%) | ||
| Paniculitis | — | — |
| Mucinosis | — | — |
| Eritrodermia | 1 (1,5) | — |
| Cambios en la mucosa oral | 1 (1,5) | — |
| Eritema flagelado | — | — |
| Manos de mecánico | 6 (9,5) | 1 (14,2) |
| Hiperqueratosis folicular | 2 (3,1) | 1 (14,2) |
| Manifestaciones dermatológicas inespecíficas, n (%) | ||
| Fenómeno de Raynaud | 10 (15,8) | 1 (14,2) |
| Prurito | 3 (4,7) | — |
| Fotosensibilidad | 14 (22,2) | 2 (28,5) |
Dentro de las manifestaciones sistémicas, 19 (30,15%) pacientes presentaron manifestaciones gastrointestinales, 12 (19,0%) manifestaciones pulmonares y 3 (4,7%) manifestaciones cardíacas. De los 63 pacientes con dermatomiositis clásica, 11 (17,4%) refirieron síntomas constitucionales, 12 (19,0%) presentaron de manera concomitante otras enfermedades del tejido conectivo y 6 (9,5%) presentaron cáncer, de los cuales 3 fueron de mama, 2 de pulmón y uno colorrectal (tabla 3).
Manifestaciones sistémicas
| Clásica | Amiopática | |
|---|---|---|
| Manifestaciones pulmonares, n (%) | ||
| Enfermedad pulmonar intersticial | 8 (12,6) | — |
| Neumonía por aspiración | 2 (3,1) | — |
| Hipertensión pulmonar | 6 (9,5) | — |
| Hipoventilación alveolar | 1 (1,5) | — |
| Neumotórax | — | — |
| Neumomediastino | — | — |
| Manifestaciones gastrointestinales, n (%) | ||
| Disfagia | 15 (23,8) | — |
| Reflujo gastroesofágico | 4 (6,34) | — |
| Retraso en el vaciamiento gástrico | - | — |
| Disminución de la motilidad intestinal | 3 (4,7) | — |
| Incontinencia rectal | — | — |
| Manifestaciones cardiacas, n (%) | ||
| Miocarditis | 1 (1,5) | — |
| Falla cardiaca | — | — |
| Defectos en la conducción | — | — |
| Pericarditis | - | — |
| Enfermedades valvulares | 2 (3,1) | — |
| Otras manifestaciones sistémicas, n (%) | ||
| Vasculitis retiniana | — | — |
| Lesión renal aguda | — | — |
| Compromiso del sistema nervioso central secundario a vasculitis | — | — |
| Síntomas constitucionales, n (%) | ||
| Fiebre | 3 (4,7) | — |
| Pérdida de peso | 11 (14,2) | 3 (42,8) |
| Asociación con malignidad, n (%) | 6 (9,5) | — |
| Asociación con enfermedades del tejido conectivo, n (%) | ||
| Lupus eritematoso sistémico | 5 (7,9) | — |
| Esclerodermia | 6 (9,5) | — |
| Síndrome de Sjögren | 1 (1,5) | — |
| Artritis reumatoide | 1 (1,5) | — |
A 25 (39,6%) pacientes les realizaron biopsia de piel, de los cuales 15 presentaron hallazgos compatibles con dermatomiositis; 24 pacientes (38,0%) fueron sometidos a biopsia muscular, 18 mostraron hallazgos acordes con esta miopatía inflamatoria (músculo esquelético con inflamación crónica perimisial, atrofia de fibras musculares y en algunos casos vacuolización) y uno fue inespecífico; 35 (55,5%) pacientes contaban con electromiografías, 25 mostraron disminución de la amplitud y la velocidad de los potenciales motores; en algunos casos se evidenciaron potenciales de fasciculación, fibrilación y ondas agudas positivas, además de descargas repetitivas complejas. Siete pacientes tenían biopsia muscular, biopsia de piel y electromiografía de forma concomitante, y tres de estos presentaron hallazgos sugestivos de dermatomiositis en los tres exámenes, de los cuales uno presentaba solo pápulas de Gottron, otro eritema heliotropo y poiquilodermia, y el último, pápulas de Gottron, eritema heliotropo, poiquilodermia y fotosensibilidad. A 33 pacientes les tomaron TAC de tórax, siete presentaron hallazgos asociados con la enfermedad, dentro de los cuales vale la pena resaltar infiltrados intersticiales, bronquiectasias, hipertensión pulmonar, tractos fibrosos, neumonía en organización y neumonía intersticial. A 16 pacientes se les tomó ecocardiograma, seis de los cuales presentaron hallazgos asociados con esta miopatía.
En cuanto a los laboratorios de inmunología, el más frecuentemente encontrado fue el de anticuerpos antinucleares (ANA), en 41 (65,0%) pacientes, siendo los patrones granular (53,6%) y homogéneo (21,9%) los más documentados. En segundo lugar, están los antígenos nucleares extraíbles (ENA), que fueron positivos en ocho (12,6%) pacientes, siendo el anti-Ro el hallado en más ocasiones. Por último, dos (3,1%) pacientes presentaron anti Jo-1 positivo.
A todos los pacientes se les solicitó creatincinasa (CK), 50 (79,3%) presentaron elevación de esta (valor de referencia: 55-170U/l). A 61 pacientes les solicitaron transaminasas, 40 (65,5%) presentaron elevación de ALT y 46 (75,4%) de AST (valor de referencia: ALT 14-54U/l y AST 15-41U/l). A 28 pacientes se les solicitó LDH, 26 (92,8%) la tuvieron elevada (valor de referencia: 120-246U/l), y de los 27 pacientes a quienes se les solicitó aldolasa, en 13 (48,1%) estaba elevada (valor de referencia: 0-7,6U/l). En cuanto a los reactantes de fase aguda, a 55 pacientes se les solicitó velocidad de sedimentación globular (VSG), que fue positiva en 46 (83,6%) caso (valor de referencia: 0-20mm/h), y a 47 pacientes se les solicitó proteína C reactiva (PCR) (valor de referencia: menor a 10mg/l), la cual resultó positiva solamente en cinco casos (10,6%). Del total de pacientes, cinco (7,9%) fallecieron, cuatro de ellos por falla respiratoria y uno por causas neurológicas (tabla 4).
Exámenes diagnósticos
| Clásica | Amiopática | |||
|---|---|---|---|---|
| Tipo de examen | Exámenes realizados | Exámenes positivos | Exámenes realizados | Exámenes positivos |
| Anti-Jo, n (%) | 33 (52,3) | 2 (6,0) | 1 (14,2) | — |
| Anti-Mi 2, n (%) | 1 (1,5) | — | — | — |
| ANA, n (%)Homogéneo, n (%)Periférico, n (%)Nucleolar, n (%)Centromérico, n (%)Citoplasmático, n (%)Granular, n (%)Ribosomal, n (%)No especificado, n (%) | 49 (77,7) | 41 (83,6)9 (21,9)—3 (7,3)1 (2,4)2 (4,8)22 (53,6)1 (2,4)3 (7,3) | 6 (85,7) | 4 (57,1)1 (25)———3 (75)— |
| Anti-SM, n (%) | 40 | — | 6 | — |
| Anti-RNP, n (%) | 40 | 3 (7,5) | 6 | — |
| Anti-La, n (%) | 40 | 1 (2,5) | 6 | — |
| Anti-Ro, n (%) | 40 | 6 (15) | 6 | — |
| CK total-VR 55-170 U/l, n (%) | 63 | 50 (79,3) | 7 | 2 (28,5) |
| ALT-VR 14-54 U/l, n (%) | 61 | 40 (65,5) | 5 | 3 (60) |
| AST-VR 15-41 U/l, n (%) | 61 | 46 (75,4) | 5 | 3 (60) |
| LDH-VR 120-246 U/l, n (%) | 28 | 26 (92,8) | 2 | 2 (100) |
| Aldolasa-VR 0-7,6 U/l, n (%) | 27 | 13 (48,14) | 5 | — |
| VSG-VR 0-20 mm/h, n (%) | 55 | 46 (83,6) | 6 | — |
| PCR-VR menor a 10 mg/ml, n (%) | 47 | 5 (10,6) | 6 | — |
| Electromiografía, n (%) | 35 | 25 (71,4) | — | — |
| Biopsia de piel, n (%) | 25 | 15 (60) | 7 | 7 (100) |
| Biopsia muscular | 24 | 18 (75) | 1 | — |
| TAC de tórax | 33 | 7 (21,2) | 1 | — |
| Ecocardiograma | 16 | 6 (35,7) | — | — |
ALT: alanina aminotransferasa; ANA: anticuerpos antinucleares; AST: aspartato aminotransferasa; CK: creatina cinasa; LDH: deshidrogenasa láctica; PCR: proteína C reactiva; TAC: tomografía axial computarizada; VR: valor de referencia; VSG: velocidad de sedimentación globular.
Sesenta (95,2%) pacientes recibieron manejo con glucocorticoides, 30 (50%) requirieron pulsos con metilprednisolona y continuaron con prednisolona oral, en tanto que 30 (47,6%) recibieron solamente prednisolona oral. En cuanto a los inmunomoduladores, los usados con mayor frecuencia fueron azatioprina, en 35 pacientes (64,8%); metotrexate, en 27 (50%); y ciclofosfamida, en 11 (20,3%) (tabla 5).
Tratamiento
| Clásica | Amiopática | Total | |
|---|---|---|---|
| Corticoides, n (%)Metilprednisolona, n (%)Deflazacort, n (%)Prednisolona, n (%) | 60 (95,2)30 (50)2 (3,3)60 (100) | 4 (57,1)1 (25)—4 (100) | 64 (92,8)31 (52,5)2 (2,8)64 (91,4) |
| Inmunomoduladores, n (%)Azatioprina, n (%)Metotrexate, n (%)Ciclofosfamida, n (%)Gammaglobulina, n (%)Cloroquina, n (%)Rituximab, n (%)Ciclosporina, n (%)Micofenolato, n (%)Tofacitinib, n (%) | 54 (85,7)35 (64,8)27 (50)11 (20,3)8 (14,8)5 (9,2)4 (7,4)3 (5,5)2 (3,7)1 (1,8) | 4 (57,1)1 (25)2 (50)——1 (25)———— | 58 (82,8)36 (51,4)29 (41,4)11 (15,7)8 (11,4)6 (8,5)4 (5,71)3 (4,28)2 (2,8)1 (1,4) |
| Un inmunomodulador, n (%) | 27 (42,8) | 4 (57,1) | 31 (53) |
| ≥ 2 inmunomoduladores, n (%) | 27 (42,8) | 0 | 27 (46) |
Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de enfermedades que afectan el sistema musculoesquelético y que usualmente se acompañan de compromiso extramuscular, debido a enfermedad pulmonar intersticial, artritis o malignidad7. Se dividen en dermatomiositis, polimiositis, miositis por cuerpos de inclusión y otras miositis, en las que se incluyen miositis eosinofílica, granulomatosa, nodular focal, infecciosa, inducida por medicamentos, orbitaria, miofascitis macrofágica, miositis osificante, miopatías hereditarias asociadas con inflamación muscular y miositis necrosante inmunomediada, entre otras1.
La forma clásica de dermatomiositis se caracteriza por la presencia de debilidad muscular proximal simétrica y manifestaciones dermatológicas; no obstante, hay una variedad en la cual la debilidad muscular está ausente, conocida como dermatomiositis amiopática7, que se presenta entre el 10 y el 20% de los pacientes8. Un estudio publicado en Italia en el 2014, que incluyó 103 pacientes con dermatomiositis, documentó la variante amiopática en el 7,7% de los pacientes; al comparar la edad de inicio de la enfermedad, los autores evidenciaron que aquellos con dermatomiositis amiopática tenían menor edad en el momento del diagnóstico9. En nuestro estudio, se reportó un porcentaje de dermatomiositis amiopática acorde con los reportes de la literatura, sin embargo, al contrario del estudio mencionado anteriormente, estos pacientes tenían mayor edad de diagnóstico, en comparación con los que tenían dermatomiositis clásica.
Se han descrito dos picos de incidencia de dermatomiositis, el primero entre los 5 y los 15 años y el segundo entre los 40 y los 60 años. Se considera que esta es una enfermedad que predomina en el sexo femenino, con una relación mujer:varón de 2:11. En cuanto a las características demográficas, en nuestro estudio el promedio de edad en el momento del diagnóstico fue similar al reportado en otro estudio, publicado en el 2012, que fue llevado a cabo en Brasil y que incluyó 139 pacientes, en el cual se reportó una edad promedio al momento del diagnóstico de 41,7 años (DS±14,1). Se han publicado hallazgos similares en otros países, como Japón, 47,4 años; México, 34 años (DS±15); Francia, 52 años; y Malasia, 57,8 años (DS±11,1)10–14. Adicionalmente, evidenciamos predominio de la enfermedad en sexo femenino, lo cual coincide con lo reportado hasta el momento en estudios realizados en Singapur, México, Francia, Taiwán y Japón11–13,15,16. En Colombia, se llevó a cabo un estudio similar al nuestro en el 2014, que incluyó a 30 pacientes con dermatomiositis y a 16 con polimiositis, con una relación mujer:varón discretamente mayor17.
En cuanto a las manifestaciones dermatológicas, en varios estudios se ha descrito que el eritema heliotropo y las pápulas de Gottron son los signos más frecuentes, lo que concuerda con nuestros resultados10,16,18,19. Los porcentajes de frecuencia de telangiectasias periungueales y calcinosis cutis tienden a ser similares a los reportados por nosotros, así como en el estudio publicado por De Souza et al.10 en Brasil, con el 6,5% de los pacientes con calcinosis cutis, y en el de Sato et al.18, también en Brasil, con el 5,3% de los pacientes con calcinosis cutis y el 38,2% con telangiectasias periungueales. Sin embargo, en nuestro estudio los pacientes presentaron menor fotosensibilidad (22,2%) y signo de funda de la pistola (17,4%), además de mayor frecuencia de signo del chal (30,1%) y signo de la V (47,6%) con respecto a lo publicado por De Souza et al.10 en Brasil, con el 15,1% de los pacientes con signo de la V y el 14,4% con signo del chal; por Sato et al.18, también en Brasil, con el 50,6% de pacientes con fotosensibilidad; por Vega et al.17 en Colombia, con signo del chal en el 16% de los pacientes y signo de funda de la pistola en el 26,6%; además de lo publicado por Hoesly et al.20 en los EE. UU., con el 22,1% de los pacientes con signo del chal.
Con respecto a las manifestaciones sistémicas y los síntomas constitucionales, la disfagia, la enfermedad pulmonar intersticial y la pérdida de peso fueron las más frecuentes. En un estudio europeo publicado en el 2011, se documentó el doble de frecuencia de enfermedad pulmonar intersticial que lo encontrado en el presente estudio, además de mayor morbimortalidad asociada en esos pacientes21. Varios estudios coinciden en que las manifestaciones gastrointestinales, pulmonares y la pérdida de peso son las más prevalentes18,20,22,23.
Como parte de las variables estudiadas, se documentó elevación de al menos una de las enzimas musculares en el 85% de los pacientes, un valor menor al encontrado en otros estudios14,18. Dentro de las enzimas musculares, la CK es la que con mayor frecuencia se encuentra elevada, los demás resultados son variables en diferentes artículos13,15,19. Generalmente, las transaminasas se elevan en proporciones similares, siendo usualmente la AST más alta que la ALT13,23.
Los ANA fueron positivos, con un porcentaje menor que el nuestro, en tres estudios brasileños: el primero, publicado por De Souza et al.10, que incluyó 139 pacientes con dermatomiositis, de los cuales el 62,6% tuvo ANAS positivos; el segundo, de Sato et al.18, que incluyó a 189 pacientes con dermatomiositis juvenil, de los cuales el 41,4% tuvo ANA positivos y, por último, un estudio por Sallum et al.24, el cual incluyó a 39 pacientes con dermatomiositis juvenil, de los cuales el 40% presentó ANA positivos10,18,24. Varios artículos han documentado mayor frecuencia de patrón de fluorescencia granular y homogéneo, lo cual concuerda con nuestros resultados10,17.
A diferencia de los resultados de un estudio brasileño publicado en el 2009, que reportó hallazgos sugestivos de dermatomiositis en el 93,2% de las electromiografías, el 91,5% de las biopsias musculares y el 77,3% de las biopsias de piel18, nosotros encontramos una menor proporción de electromiografías (71,4%), lo que concuerda con un estudio realizado en Singapur, en el que se reportó un 79,4% de pacientes con electromiografías15.
En cuanto al tratamiento, el 95,2% de nuestros pacientes recibió manejo con glucocorticoide oral o intravenoso; hasta el momento se han documentado proporciones similares en Asia, México y Brasil10,11,13,14. Con referencia a la terapia de mantenimiento, la mayoría de los pacientes en nuestro estudio recibió al menos un inmunomodulador; dentro de los inmunomoduladores, los utilizados con mayor frecuencia, de acuerdo con un estudio llevado a cabo en Brasil que incluyó a 139 pacientes, son azatioprina y metotrexate10, lo cual coincide con nuestros resultados.
En nuestro estudio se evidenció, de manera concomitante, enfermedades del tejido conectivo en el 17% de los pacientes, lo que concuerda con datos documentados en Asia15. La mayoría de las enfermedades concomitantes fueron lupus y esclerosis sistémica; no se han documentado hasta el momento estudios que muestren estas dos entidades de forma predominante, sin embargo, en un estudio llevado a cabo en Brasil, publicado en el año 2000, en el que se incluyeron 59 pacientes con dermatomiositis y 43 pacientes con polimiositis, en el grupo de pacientes con dermatomiositis se documentó esclerosis sistémica en siete pacientes y artritis reumatoide en uno22. Además, en un estudio publicado en Singapur, que incluyó a 40 pacientes con dermatomiositis y a 35 con polimiositis, 10 de ellos presentaron asociación con lupus eritematoso sistémico, cuatro con artritis reumatoide y uno con esclerosis sistémica; en total, el 21,4% de los pacientes presentó de forma concomitante otra enfermedad del tejido conectivo. Por último, en el estudio publicado por Sato et al., en el que se incluyeron 189 pacientes, ocho presentaron artritis reumatoide, dos lupus eritematosos sistémico y uno esclerosis sistémica15,18.
Se estima que aproximadamente el 15% de los pacientes con dermatomiositis diagnosticada después de los 40 años va a presentar una neoplasia tres a cinco años después del diagnóstico, siendo las más comunes los carcinomas colorrectal, ovárico, pulmonar, pancreático y gástrico25. En un estudio realizado en Estocolmo, Suecia, publicado en 1992, que incluyó a 392 pacientes con dermatomiositis, se documentó un riesgo relativo de cáncer en varones de 2,4 (IC 95%: 1,6-3,6) y en mujeres de 3,4 (IC 95%: 2,4-4,7)26. En nuestro estudio se encontró un porcentaje similar de pacientes con cáncer a lo reportado por los estudios de Scola et al.22 en Brasil (8,4%) y Chen et al.23 en China (11,1%). Por el contrario, otros estudios han reportado porcentajes mayores de malignidad, como los de Hiketa et al.13 en Japón (16,5%), Fardet et al.12 en Francia (23%) y Hoesly et al.20 en los EE. UU. (23%).
Con respecto a la mortalidad, en un estudio publicado en el 2018, que incluyó a 982 pacientes hospitalizados con diagnóstico de dermatomiositis y polimiositis en China, se documentó el fallecimiento de 63 de ellos durante la hospitalización o dos semanas después de haberles dado de alta, con una tasa de mortalidad calculada de 6,4%, siendo las dos causas más frecuentes de muerte infección pulmonar y exacerbación de la enfermedad pulmonar intersticial27, lo que se asemeja a nuestros resultados. Sin embargo, en un estudio retrospectivo en EE. UU., que incluyó a 15.407 pacientes (10.023 con diagnóstico de dermatomiositis y 5.384 de polimiositis), se encontró una tasa de mortalidad de 4,4% en el grupo con diagnóstico de dermatomiositis (449 muertes), porcentaje menor al nuestro28.
Dado que nuestro estudio recolectó los datos en forma retrospectiva, dentro de las limitaciones es importante mencionar que no se pudo obtener datos de seguimiento, con el fin de determinar el pronóstico de los pacientes. Los datos fueron tomados de registros médicos electrónicos, lo que puede limitar su recolección, en especial los que hacen referencia a las características clínicas. No a todos los pacientes les realizaron los mismos paraclínicos, sin embargo, es un reflejo de la práctica clínica, en la cual la solicitud de pruebas diagnósticas está en relación con el cuadro clínico de los pacientes. Finalmente, cabe resaltar que este es hasta el momento el estudio con mayor número de pacientes con dermatomiositis en Colombia.
ConclusiónSe presentan las principales características demográficas, las manifestaciones dermatológicas y sistémicas de un grupo de pacientes con dermatomiositis en un hospital universitario en Colombia. La mayoría de los datos obtenidos concuerda con la información de otros estudios de series de casos a escala mundial. A pesar de que el eritema heliotropo, las pápulas y el signo de Gottron se han definido como manifestaciones patognomónicas, de conformidad con lo propuesto en las revisiones narrativas por DeWane et al. y Mainetti et al.1,6, en nuestro estudio encontramos que las manifestaciones más frecuentes son pápulas de Gottron, eritema heliotropo y poiquilodermia. La población evaluada presentó en menor proporción fotosensibilidad y signo de funda de la pistola, además de mayor frecuencia de signo del chal y signo de la V, en comparación con otros estudios.
Aprobación éticaAprobación por el Comité de Investigaciones y Ética Institucional de la Facultad de Medicina de la Pontificia Universidad Javeriana – Código 2019/207 – Número de Acta 18/2019.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses para la elaboración de este artículo.
