




The case is presented on a 52-year-old male patient, who was seen in the Rheumatology department. He had painless lymph nodes in the cervical, axillary, supraclavicular, and neck region. He also had a fever, and parotid and submaxillary gland enlargement. Complementary studies were performed, showing normocytic-normochromic anemia, thrombocytopenia and eosinophilia, impaired renal function with hypoalbuminaemia and hematuria, ANA 1/5120, Sm+, ACL+. Biopsies were also performed on the compromised tissues, reaching the diagnosis of Rosai-Dorfman Disease and IgG4-related Disease. Differential diagnoses of cervical, axillary and inguinal lymph nodes, with fever, renal and hematological compromise are discussed.
Se describe el caso de un paciente varón de 52 años que consulta al servicio de reumatología por presentar adenopatías indoloras en región cervical, axilar, supraclaviculares y en la nuca, así como fiebre, aumento de tamaño de parótidas y submaxilares. Se realizan estudios complementarios que arrojan como resultado anemia normocitica-normocrómica, plaquetopenia y eosinofilia, alteración de la función renal con hipoalbuminemia y hematuria, FAN 1/5120, sm+, ACL+ y biopsia de los tejidos comprometidos, por lo que se arriba al diagnóstico de enfermedad de Rosai Dorfman y enfermedad relacionada con IgG4. Se discuten diagnósticos diferenciales de adenopatías cervicales, axilares e inguinales, fiebre, compromiso renal y hematológico.
Rosai-Dorfman disease (RDD) is a rare entity characterized by fever, lymphadenopathy, neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Anatomopathological findings are characterized by histiocytic infiltration and other inflammatory cells, together with emperipolesis. Immunohistochemically, it shows histiocytes positive for S100 and CD68; likewise, it is characteristic that histiocytes are negative for CD1a. Extranodal disease, reported in 33%–44% of cases, is located more frequently in the head and neck, while renal involvement has been described in 4% of cases, associated with a worse prognosis and a mortality rate of 40%.
IgG4-related disease (IgG4-RD) is a new clinical entity, characterized by an elevated serum concentration of IgG4 and tissue swelling or infiltration by IgG4-positive plasma cells, accompanied by storiform fibrosis. It can manifest similarly to Mikulicz disease, autoimmune pancreatitis, hypophysitis, Riedel's thyroiditis, interstitial pneumonitis, interstitial nephritis, prostatitis, lymphadenopathy, retroperitoneal fibrosis, inflammatory aortic aneurysm and inflammatory pseudotumor.
In the literature there are few case reports on the association of these two entities, which share clinical and histological characteristics. We present the case of a 52 year-old man with multiple painless cervical adenopathies, with renal and salivary glands involvement, which constituted a diagnostic challenge. It is considered opportune to take into account these little-known diseases when making a correct diagnosis.
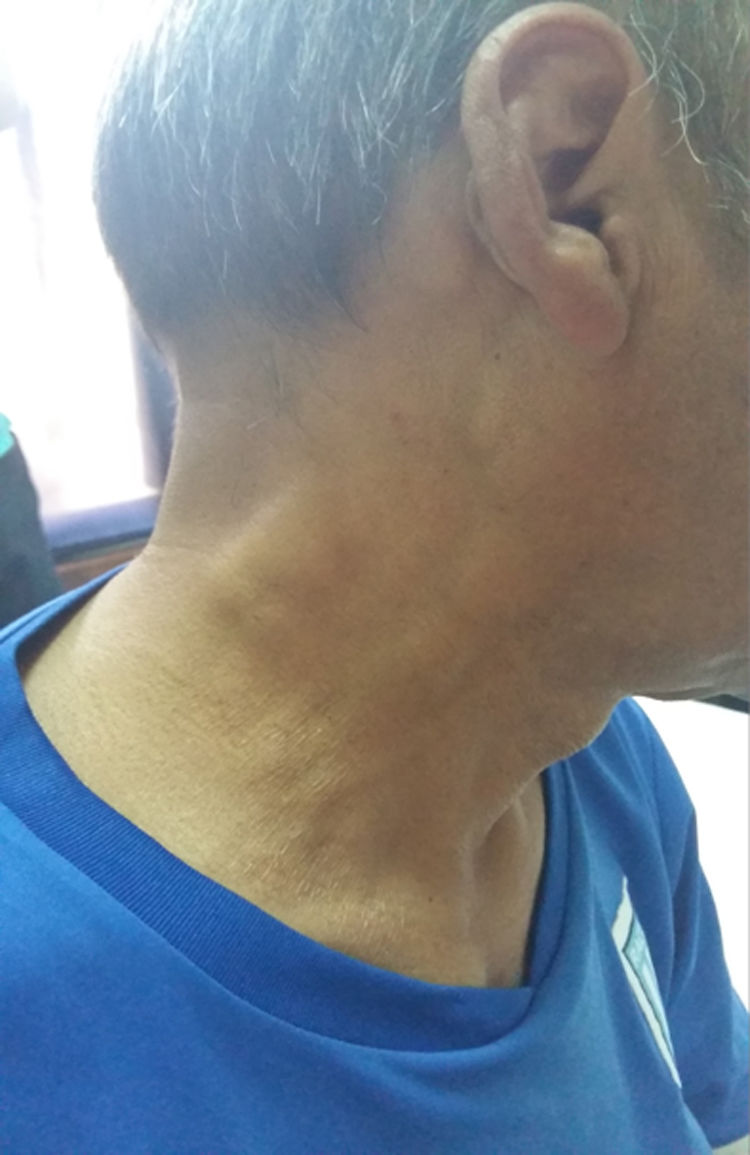
Case presentationA 52-year-old male patient with a history of type 2 diabetes mellitus who consulted for fever, painless bilateral adenopathies in the cervical region, back of the neck, axilla, and supraclavicular regions (Fig. 1). Enlargement of the salivary glands: parotids and submandibular was also found (Fig. 2). No visual changes, sicca symptoms, malar rash, Raynaud's phenomenon, nasal or oral ulcers, hair loss, dysphagia, dyspnea or cough were observed. There was also no family history of neurological disorders or thyroid diseases. The patient denied toxic habits, use of illegal drugs or allergy to drugs. The physical examination revealed fever, bilateral cervical, axillary, and inguinal lymphadenopathies, of 2−3 cm in diameter. No alterations were found in the cardiovascular and pulmonary examination. The abdomen was found to be normal.


In the laboratory, the patient exhibited normochromic normocytic anemia, thrombocytopenia and eosinophilia. Likewise, increased erythrocyte sedimentation rate and CRP; decreased C3 and C4; polyclonal hypergammaglobulinemia; proteinuria 1518 mg (30−140 mg/24 h); blood creatinine 4.15 mg/dL (normal range = 0.50−0.90 mg/dL); urea 1.18 g/l (normal range = 0.10−0.40 g/l); hypoalbuminemia 1.5 g/dL (normal range = 3.50–4.80 g/dL) and hematuria; positive antinuclear factor (ANF hep2) 1/5220 (histone) and anticardiolipin (ACL) antibodies; negative lupus anticoagulant and beta 2 glycoprotein; elevated serum IgG and IgE; the rheumatoid factor, anti-DNA ds, anti-Ro; La, RNP and P and C ANCA ELISA for PR3 and myeloperoxidase were negative; normal antithyroid antibodies and thyroid hormones; serum IgG 4 and IgG 1376 mg/dL (700–1600 mg/dL) and 948 mg/l (110−1570 mg/l), respectively. The blood and urine cultures were negative. The evaluations for syphilis, human immunodeficiency virus, tuberculosis, toxoplasmosis, mycoplasma, cytomegalovirus, Epstein-Barr virus, hepatitis B and C viruses were negative.
A renal biopsy was performed, whose result reported renal interstitial infiltration by lymphohistiocytic elements, glomerulus with slight proliferation of mesangial cells, 20% tubular atrophy, and interstitial nephritis. The patient presented interstitial fibrosis (not storiform) and lymphoplasmacytic infiltrates (predominantly lymphocytes) without eosinophils. Obliterative phlebitis was not observed. Immunolabeling: 15 IgG4+ cells per high field, IgG4/IgG ratio of 25%. A biopsy of a cervical lymph node was also performed, which presented lymphoid follicles with germinal centers. The interfollicular areas were expanded, at the expense of numerous plasma cells and foci of emperipolesis. Immunolabeling: positive CD68, positive S100, negative CD1a, IgG4 100 cells per high field and an IgG4/IgG ratio of 45%.
The bone marrow biopsy revealed hypercellularity with a myelolipomatous ratio of 80/20, increase in plasma cells of 50%, CD138+ and foci of emperipolesis. The minor salivary gland biopsy showed lymphoplasmacytic infiltration, mild interstitial fibrosis (not storiform), without obliterative phlebitis or eosinophils. Periductal lymphocytic accumulations. Immunolabeling, IgG4: 12 cells per high field and an IgG4/IgG range of 25%. Despite presenting these antibodies, it does not meet the diagnostic criteria for systemic lupus erythematosus (SLE), according to the ACR 1997 criteria.
Material and methodA bibliographic search was carried out in rheumatology books (Robert G. Lahita, George Tsokos, Jill Buyon, Takao Koike; Systemic Lupus Erythematosus. Fifth Edition. USA. Elsevier, 2011) and a systematic search of bibliography in databases (MEDLINE, PubMed, LILACS) using terms in Spanish and English such as “adenopathy”, “IgG4-related disease”, “histiocytosis” and “systemic lupus erythematosus”, without deadlines for the results.
Differential diagnosisThe differential diagnosis of a patient with cervical, axillary and inguinal lymphadenopathies, fever and renal and hematological involvement includes: SLE, primary Sjögren's syndrome (pSS), IgG4-RD, Castleman disease, Kikuchi-Fujimoto disease, PFAPA syndrome (periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis) and RDD.
Systemic lupus erythematosusIn SLE, lymphadenopathy occurs in 50% of all patients during the course of the disease. The most compromised groups are cervical (43%), mesenteric (21%), axillary (18%) and inguinal (17%). Cervical lymphadenopathy is the first clinical manifestation of the disease in 2% of patients. The adenopathies are typically non-inflammatory, soft and discrete, they vary in size—between one and several centimeters—and occur in young patients. Histologically, the lymph nodes show paracortical foci of necrosis and infiltration of lymphocytes, histiocytes, plasma cells, and immunoblasts, with “hematoxylin bodies” that are classic of lupus lymphadenopathy. Fever is a common sign of SLE activity, but infection must always be considered. For this reason, this presumptive diagnosis was excluded, since the histology of our patient did not show said reactive hyperplasia or hematoxylin bodies, nor did it meet the 1982 ACR classification criteria for SLE.
Other important diseases, such as RDD, Castleman disease, lymphoma, and Kikuchi disease have been associated with lymphadenopathies and SLE. Histiocytic necrotizing lymphadenitis of SLE is pathologically similar to Kikuchi-Fujimoto disease. These two clinical entities can also coexist, or Kikuchi disease can precede the diagnosis of SLE. Castleman disease or angiofollicular lymph node hyperplasia is a rare disease of unknown etiology that can present as local or multicentric lymphadenopathy. This multicentric lymphadenopathy and other clinical characteristics can mimic SLE.1
Kikuchi-Fujimoto diseaseKikuchi-Fujimoto disease, also known as necrotizing histiocytic lymphadenitis, is a self-limited disease of young women, of unknown cause, which can mimic SLE. The signs and symptoms include cervical lymphadenopathy, fever, weight loss, and a prodrome of upper respiratory tract infection. The diagnosis is based on the histological findings of the biopsy. Cervical adenopathies are the main site of the disease and are seen in 70%–98% of patients. However, involvement of other nodal areas has been described, including peripheral (mainly axillary) thoracic, abdominal, and pelvic lymphadenopathies. Fever is a primary symptom in 30%–50% of the cases.
Although certain clinical characteristics are commonly present in this entity (cervical lymphadenopathy, leukopenia, elevated erythrocyte sedimentation rate, and age less than 30 years), there is nothing specific that suggest it.2 Histologically, it is characterized by paracortical expansion of the lymph nodes with well-circumscribed, irregular areas of necrosis showing abundant karyorrhectic nuclear remnants and the absence of neutrophils and eosinophils. Three evolving histologic patterns have been recognized: proliferative, necrotizing, and xanthomatous. By immunohistochemistry, the histiocytes are positive for myeloperoxidase. There is a marked predominance of T cells in the lesions (with mostly CD8-positive cells) and very few B cells.1
The differential histological diagnosis of Kikuchi disease includes other causes of necrotizing lymphadenitis, specifically infections, autoimmune and neoplastic diseases. The biopsy is important to exclude malignancy and confirm the diagnosis.2 Although our patient is a man and presented clinical and laboratory findings compatible with Kikuchi-Fujimoto disease, when reviewing the histological involvement and the immunohistochemistry, these findings did not turn out to be characteristic of this disease.
Sjogren's syndromeAnother diagnosis that must be taken into account is SS, an autoimmune exocrinopathy characterized by dry eye, dry mouth, and parotid enlargement. The most worrisome complication is the development of a lymphoproliferative disorder, most commonly a non-Hodgkin's lymphoma. Lymphadenopathy is a frequent sign of this disease. Patients may present with a single enlargement, or in multiple sites, but massive lymphadenopathy, with or without splenomegaly, in SSs is a warning sign of the development of a lymphoproliferative disorder. Likewise, the lymphadenopathy associated with a lymphoproliferative disorder requires the presence of neoplastic lymphoid cells in the bone marrow or lymph node biopsy.
Drosos et al. describe a male patient with SS who developed RDD. The patient had been admitted to the hospital with dry mouth, bilateral parotid enlargement, generalized lymphadenopathies and splenomegaly.3 The minor salivary gland biopsy showed focal sialadenitis with a score of one focus according to the Chisholm and Mason criteria, being the first case published in the literature with this association. The patient had developed an infection with Brucella, then developed SS and shortly thereafter a sinus histiocytosis with massive lymphadenopathy (SHML). It is speculated that a chronic infection such as brucellosis and an immune disorder such as SS may lead to the development of SHML. An association of these two entities cannot be excluded either.4 Although sicca symptoms were not found in our patient, when performing a biopsy of the gland, no characteristic histological pattern of SS was found either.
Antiphospholipid syndromeAntiphospholipid antibodies, present in this patient (for a single time), have been reported in patients with lupus nephritis and in those without other characteristics of systemic disease (e.g., antiphospholipid syndrome). Despite the presence of high titers of ACL in the patient, he did not present clinical characteristics of antiphospholipid syndrome and he has only one serum dosage of ACL and lupus anticoagulant. The patient had many clinical manifestations of SLE: generalized lymphadenopathy, unexplained fever, and weight loss. The diagnosis is confirmed by the presence of antibodies in serum. Although ANF can be seen in other autoimmune diseases, anti-native DNA and anti-SM antibodies are relatively specific for the diagnosis.
The cause of acute renal failure associated with nephritic syndrome is less obvious. Although such a cause is usually apparent, this unexplained failure is an indication for renal biopsy. Interstitial nephritis cannot be excluded, due to the association of pyuria with acute renal failure. Interstitial involvement is common in lupus nephritis, and extensive interstitial fibrosis is closely related to progressive renal failure. The non-steroidal anti-inflammatory drugs—received by this patient—can cause interstitial nephritis. We also considered an allergic interstitial nephritis due to the cephalosporin that the patient had received, acute tubular necrosis based on hypoalbuminemia, and antiphospholipid syndrome.1
InfectionsWithin the spectrum of infectious diseases, tuberculosis, yersiniosis, toxoplasmosis, cat scratch disease, and lymphogranuloma venereum, among others, must be ruled out. Localized infection is the most frequent cause of fever and cervical lymphadenopathy. The most common form of localized infection is pharyngitis, which can be viral, caused by mycoplasma, or pyogenic and due to streptococcus, staphylococcus aureus and staphylococcus epidermidis. Tonsillitis due to a variety of organisms can present as cervical lymphadenopathy. Tuberculous cervical lymphadenitis (scrofula) is historically the most common cause of extrapulmonary tuberculosis.1 Even though these are always taken into account as possible causes, they were excluded due to negative serologies and cultures.
Rosai-Dorfman diseaseSinus histiocytosis with massive lymphadenopathy (SHML), also known as Rosai-Dorfman disease (RDD), is a rare histiocytic proliferative disorder of unknown etiology, initially described by Rosai and Dorfman in 1969.5 The World Health Organization (WHO) and the Histiocyte Society include it in group II of the Classification of Histiocytosis and Dendritic Cell Diseases, which encompasses non-Langerhans cell mononuclear phagocyte histiocytosis, as opposed to Langerhans cell histiocytosis (group I) and malignant histiocytosis (group III).6
RDD is a sinus histiocytosis with massive lymphadenopathy that predominantly affects children. It also presents massive bilateral infiltrates in the orbital soft tissue and extensive cervical lymphadenopathy. Minimal forms that affect the eyelids and the conjunctiva can occur in adults. It is characterized by painless lymphadenopathy with fever, leukocytosis, neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. The most frequent form of presentation of this entity consists of massive and painless bilateral cervical lymphadenopathies.7 As can be seen, those presented by our patient, accompanied by the laboratory results, had interstitial renal and salivary gland involvement.
The etiologic hypothesis of RDD includes disorders of immune regulation, as well as infections caused by agents such as herpesvirus, Epstein-Barr virus, cytomegalovirus, Brucella, and Klebsiella. Immunological abnormalities and their significance in RDD have been reported by Foucar et al. in 230 cases of SHML. Of these cases, 23 were identified with clinical and laboratory findings suggestive of an immune disorder. Likewise, they found 9 patients with autoantibodies, 3 with glomerulonephritis, 2 with Wiskott-Aldrich syndrome, 9 with musculoskeletal manifestations, 3 with unusual infections and 6 with muscular alterations.4
SarcoidosisIt is a multisystemic disorder of unknown etiology that most frequently affects adults between 20 and 40 years of age. The patients with sarcoidosis usually present bilateral hilar lymphadenopathy, pulmonary infiltration, and often ocular and skin lesions. A diagnosis of this entity is reached when the clinical and radiographic findings are supported by histological evidence of non-caseating epithelioid cell granulomas found on the tissue biopsy. A large number of patients (40%–50%) with chronic sarcoidosis have extrapulmonary involvement that can affect almost any tissue. Sarcoid granulomas are commonly infiltrated into the lymph nodes and the salivary glands.
When the hilar lymph nodes are included, the incidence of lymphadenopathy in patients with sarcoidosis probably approaches 90%, while the incidence of peripheral lymphadenopathy is about 27%. The lymphadenopathy can be associated with acute and chronic diseases and is usually painless. Cervical lymphadenopathy is the most common manifestation of sarcoidosis of the head and the neck, but sarcoidosis only accounts for 1.7% of all head and neck lymphadenopathies.6 The activity of the serum angiotensin-converting enzyme (ACE) does not appear to increase the predictive value of pulmonary function tests and chest radiographs and its use is not recommended in investigation of suspected sarcoidosis.7 Our patient did not have lung disease, skin rashes (e.g., erythema nodosum and lupus pernio), musculoskeletal complaints, or neurological dysfunction (facial nerve palsy, headache, and encephalopathy). In the same way, his ACE was negative, so it was moved away from the diagnosis.
IgG4-related diseaseIgG4-RD is a new clinical entity characterized by an elevated serum concentration of IgG4 and tissue swelling or infiltration by IgG4-positive plasma cells. It can manifest with a wide variety of entities, namely: Mikulicz disease, autoimmune pancreatitis, hypophysitis, Riedel's thyroiditis, interstitial pneumonitis, interstitial nephritis, prostatitis, lymphadenopathy, retroperitoneal fibrosis, inflammatory aortic aneurysm, and inflammatory pseudotumor.8
The concomitant lymphadenopathy, common in this disease, generally takes 2 forms. First, through the participation of the lymph nodes as a localized disease adjacent to a specific organ affected by IgG4-RD. Second, the generalized lymphadenopathy may be the sole component of the clinical presentations in IgG4-RD.
Apparently, the histomorphologic findings of IgG4-related lymphadenopathy showed histological diversity. In addition, from the clinical point of view, this entity only occasionally showed systemic lymphadenopathy, polyclonal hypergammaglobulinemia, particularly elevation of IgG and IgE, and positivity for several autoantibodies. The disease in question can be characterized into 5 histological subtypes: morphology similar to Castleman's disease (I), reactive follicular hyperplasia (II), interfollicular plasmacytosis and immunoblastosis (III), progressive transformation of the germinal center (IV) and inflammatory pseudotumor (V).9
Patients with IgG4-related systemic lymphadenopathy were significantly older (68.8 vs. 43.3 years) and they had appreciably lower concentrations of C-reactive protein (0.29 vs. 8.71 mg/dL) and interleukin (IL)-6 (8.45 vs. 34.82 pg/mL) than patients with multicentric Castleman’s disease.
One of the most important differential diagnoses is the atypical lymphoplasmacytic and immunoblastic proliferation (autoimmune disease-associated lymphadenopathy). Koo et al. reported an unusual lymph node lesion, “ALPIB”, associated with several autoimmune diseases, including rheumatoid arthritis and SLE. Histologically, it is characterized by a prominent polyclonal lymphoplasmacytic infiltration with several numbers of immunoblasts. However, there is no evidence of definite autoimmune disease in any of the IgG4-related lymphadenopathies. Atypical or malignant reactive lymphoproliferative disorders (LPD) occurring in patients immunosuppressed with methotrexate (MTX) for the treatment of rheumatoid arthritis have been reported since the early 1990s. A percentage of MTX-induced lymphadenopathy appears to be similar to ALPIB.10
When angioimmunoblastic T-cell lymphomas (AITL) contain some tumor cells (clear cells), with numerous plasma cells and B-immunoblasts, they can be confused with type III lesions. Unlike AITL, there are no cytologically atypical T CD10+ cells and no extrafollicular dendritic follicular proliferation occurs in the type III lesions. In addition, AITL often involves systemic symptoms.
Although some cases of lymphadenopathy were previously designated as atypical lymphoproliferative disorders mimicking malignant lymphomas, they lack monoclonality of the immunoglobulin gene and are considered to be non-neoplastic. Patients with IgG4-related lymphadenopathy should have lymph node biopsies to exclude lymphoma, sarcoidosis, multicentric Castleman's disease, or disseminated neoplasms.11
PFAPA syndromeThe syndrome of cervical adenitis, aphthous stomatitis, periodic fever, adult-onset pharyngitis (PFAPA), along with other entities such as Behcet's syndrome, recurrent idiopathic pericarditis, adult-onset Still's disease, systemic-onset juvenile idiopathic arthritis, belongs to the group of acquired autoinflammatory diseases (AID) on a multifactorial or polygenic basis. The acronym PFAPA (periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis) was coined together with the diagnostic criteria of 1989, 2 years after the first description of this entity made by Marshall in 1987. This entity is characterized by high fever (>39 °C) associated with at least one of the 3 cardinal clinical signs: aphthous stomatitis, pharyngitis, or cervical adenitis. It has been well described in pediatric patients, generally before the age of 5 years, and is the most frequent cause of periodic fever of unknown origin in children.
About 40 cases of adult-onset have been described in the literature. The clinical characteristics of adult patients are similar to those described in children, except that arthralgias and myalgias appear to be more frequent in adults than in children, whereas aphthae and chills appear to be less common in adults. The 3 cardinal signs (aphthosis, pharyngitis, and cervical adenitis) were concurrently present during the attacks in 47% of adults, in contrast to 27% of children. At least 2 cardinal signs were seen in 52% of adult patients, compared to 46% of children. Likewise, the mean number of episodes/year was 8.3 ± 5.2 in the adults and 13.3 ± 9.2 in the PFAPA population. This differential diagnosis was taken because the patient presented cervical adenopathies and fever, although he did not meet all the inclusion criteria.
There are case reports on the combination of PFAPA syndrome with autoimmune manifestations such as endocapillary proliferative glomerulonephritis, type 2 autoimmune hepatitis, recurrent acute aseptic encephalitis, and acute parapsoriasis. These associations could be occasional; until now, there are no data to establish whether deregulation in PFAPA could predispose these patients to autoimmunity. There are no diagnostic tests for this syndrome. During the attacks, patients may have moderate leukocytosis with a predominance of neutrophils. The erythrocyte sedimentation rate and the C-reactive protein are constantly increased during the febrile attacks.12 In this patient, however, it was ruled out due to the absence of aphthae or pharyngitis and the presence of leukocytosis with neutrophilia.
Deficiency of adenosine deaminase 2The deficiency of adenosine deaminase 2 (DADA 2) is an autosomal recessive disease described in 2014. Initially recognized as a syndrome that manifests with fever, polyarteritis nodosa, livedo racemosa, early-onset stroke, and minimal immunodeficiency. The clinical presentation and the age of onset vary widely, and the most severe manifestations are bone marrow aplasia, pure red cell aplasia, neutropenia, liver disease, and neurological damage. Its clinical presentation is highly variable. The onset of the disease usually occurs in childhood, although it has been reported in adults. The oldest patient was 59 years old and presented with leg ulcerations. Lymphoproliferation is another important feature of this entity.
In the majority of cases, the patients present with generalized lymphadenopathy (>10%) and splenomegaly (>30%). Cases with a phenotype similar to autoimmune lymphoproliferative syndrome (ALPS) and another patient with a Castleman-like disease, associated with fever, splenomegaly, generalized lymphadenopathy, anemia, thrombocytosis, polyclonal hypergammaglobulinemia, and IL-6 have recently been described.13 This diagnosis was ruled out in this patient due to the age and the lack of vascular and neurological commitment, even though he had hematological involvement.
Kimura diseaseKimura disease is an inflammatory disorder of unknown etiology that mainly affects people of Asian origin.14 From the clinical point of view, it is usually manifested by the presence of masses of subcutaneous and subplastimal tissue, generally located in the regions of the head and neck (76%), with the participation of salivary glands and regional lymphadenopathies, as could be seen in our case. Histologically, it is characterized by involvement of the lymph nodes in the form of fibroinflammatory pseudotumors, with hyperplasia of germinal follicles, interstitial fibrosis, marked eosinophilic infiltration of the mass and the adjacent tissues, and, frequently, eosinophilia with increased IgE in the blood. It is also noteworthy that it shares with this entity histological features that are found in the cervical lymph node biopsy, but not the characteristic follicle hyperplasia or eosinophilia, nor subcutaneous masses, so the disease in question was ruled out.
Clinical diagnosis of the speakerOur patient presents the clinical characteristics of an RDD, since bilateral and massive, painless cervical lymphadenopathies were found, accompanied by fever, leukocytosis, polyclonal gammopathy and accelerated erythrocyte sedimentation rate, with extranodal involvement (bone marrow and kidney commitment). Histologically, the patient presented a characteristic of the disease, as it is emperipolesis and CD68+, S100+ and CD1a− immunolabeling. In addition, the histology of the lymph nodes showed lymphoid follicles with germinal centers, numerous plasma cells with IgG4+ cells, as well as in the kidney, bone marrow and minor salivary gland. Therefore, having ruled out other causes of cervical lymphadenopathy, it was assumed that the patient had an RDD and an associated autoimmune pathology (IgG4-RD).
Anatomopathological discussionThe lymph node biopsy revealed mainly nonspecific morphological findings, commonly associated with reactive lymphadenopathies, such as follicular hyperplasia or expansion of the interfollicular area. Due to the expansion of the interfollicular area at the expense of plasma cells, immunohistochemical techniques for IgG and IgG4 were used, and a number of IgG4+ plasma cells greater than 100 cells per high-power field was confirmed, as well as an IgG4/IgG ratio of 45%. Both indicators support the diagnosis of IgG4-RD. IgG4-RD in the lymph node behaves like a reactive lymphadenopathy with different types of presentation according to the architecture; in this case it could have been of type II (similar to reactive follicular hyperplasia) or type III (similar to interfollicular expansion).15 However, the presence of subcapsular and medullary sinuses containing CD68+, S100+ and CD1a− histiocytes with emperipolesis confirms the diagnosis of RDD.16
On the other hand, the histological findings made on both the salivary gland biopsy and the renal biopsy are relatively nonspecific and do not invalidate the diagnosis of RDD. Although in both cases more than 10 IgG4+ plasma cells are observed, the IgG4/IgG ratio is less than 40%, so the diagnosis of IgG4-RD is dismissed. Since both RDD and IgG4-RD can affect multiple organs and may be associated with autoimmune diseases, the diagnosis can be difficult. This is especially true in those cases of RDD with an increase in IgG4+ plasma cells, since around 30% of patients with RDD present more than 10 IgG4+ plasma cells per high-power field and an IgG4/IgG ratio greater than 40%, which is why certain findings overlap.17
The distinction between both diseases is important because IgG4-RD has high response rates to corticosteroids, while in RDD the response is rather low in those cases that not succeed to self-limit.16 Some authors pose the hypothesis that corticosteroid-responsive RDD cases could be those with an increased number of IgG4+ plasma cells, but there are no studies to demonstrate this.17
Unlike the IgG4-RD, the RDD does not show steriform fibrosis or obliterative phlebitis, but shows S100+ histiocytes. There are discrepancies between the biological bases that differentiate one disease from the other. Thus, some authors point out that these are different diseases since the number of FOXP3+ regulatory T lymphocytes is significantly higher in IgG4-RD. However, other authors reach contradictory results, since they observe the same phenomenon in cases of extranodal RDD, but not in those that affect the lymph node.18
Comments and definitive resultAlthough RDD can occur at any age, it is more common in children and young adults, with a slight male predominance. However, the age at which the cases reported with renal involvement occurred (4%) was clearly higher, the majority within the range between the fourth and ninth decade of life. The etiology is unknown. Alterations of the immune system or infectious agents, such as agents similar to herpes virus, Epstein-Barr virus, cytomegalovirus, Brucella or Klebsiella have been described.
RDD has a course that is characterized by remissions and exacerbations in the majority of patients. However, those who present immunological abnormalities before or after the manifestation of the disease have a less favorable prognosis and a higher mortality.
The histopathology of the compromised tissues shows emperipolesis, a distinctive characteristic of this entity. The histiocytes are CD1a negative but S100 positive.19. Some cases of RDD show many IgG4-positive plasma cells, which produces diagnostic confusion with IgG4-RD. With the classic characteristics of massive lymphadenopathies and the evidence of sinus histiocytosis, as well as the histopathological characteristics observed in the lymph node, the bone marrow and the renal lesion, a diagnosis of RDD is made. Although the absence of histological examination prevents to ensure it, it is very probable that the splenomegaly is due to histiocytic infiltration.
Although the classic RDD occurs with massive, bilateral and painless cervical lymphadenopathies, approximately 40% of the cases of RDD have extranodal involvement or in other tissues, in some cases without associated lymphadenopathy, which may or may not develop during the subsequent course of the disease. The most frequently affected extranodal sites are the skin and the soft tissues, the upper respiratory tract and the bones, followed by the genitourinary tract, the lower respiratory tract, the eyes, the pancreas and the oral cavity. In RDD with renal involvement, it is necessary to make a differential diagnosis with malignant fibrous histiocytoma and histiocytic proliferations of infectious etiology (the presence of S100 is useful to differentiate it from these lesions), leukemias and lymphomas, especially when they are accompanied by lymphadenopathies (the absence of emperipolesis and the immunohistochemical profile help to establish a correct diagnosis).
The diagnosis can be difficult and delayed due to misinterpretation of clinical and histological data, it is really challenging and can be wrong and indicate sarcoidosis, juvenile idiopathic arthritis, spondyloarthropathy or metastatic subcutaneous nodules. Other pathologies with which the differential diagnosis must be established are deposit diseases, tuberculosis, renal carcinoma and renal metastases, especially melanoma.20 The diagnosis of the disease in this case was made by clinical symptoms, histology and immunohistochemical analysis of the lymph node.
In general, RDD has a good prognosis, although its course is unpredictable and can range from spontaneous remission to persistent disease with episodes of remission and exacerbation. Between 5 and 11% of patients die as a consequence of the disease, almost always they are cases with associated immunological alterations (autoimmune hemolytic anemia, glomerulonephritis, positive RF or ANF). Renal involvement has been associated with a worse prognosis, with a mortality rate of approximately 40%. Extranodal, renal, of the liver and the lower respiratory tract involvement is associated with a worse prognosis, as well as with the number of affected extranodal organs.21 The most common extranodal manifestation or the most affected areas are the head and the neck in 22% of the cases. The orbit or the eyelids are affected in 10% of the cases.
Anemia and leukocytosis can be found in the laboratory tests. 90% of the patients have increased erythrocyte sedimentation rate and polyclonal hypergammaglobulinemia. Less frequently, antinuclear antibodies are detected. Approximately 13% of patients have concomitant autoimmune alterations, such as lupus erythematosus, rheumatoid arthritis, and autoimmune hemolytic anemia.22 The latter, the most frequent finding, in some cases is fatal.23
There are 2 reports in the literature that describe RDD in patients with SLE. We present the case of a patient with SLE and antiphospholipid antibody syndrome who had involvement of the lacrimal gland, the orbit, and nodal and extranodal compromise related to RDD. The association of RDD with SLE is very rare; there are only 3 cases reported in the literature.24 Juskevicius and Finley reported the case of a 48-year-old man with SLE and persistent enlargement of the right parotid gland. The histology was diagnostic of RDD.25
Petschner et al. describe the case of a 59-year-old woman with SLE who presented with polyarthralgia, myalgia, photosensitivity, polyserositis, and cervical, axillary, and inguinal lymphadenopathies. This patient had ANF, low complement and ACL IgM.26 As has been commented, emperipolesis is characteristic of this disease, but it can be found in other diseases, such as 1) hematologic malignancies: acute leukemia, chronic myeloid leukemia, Hodgkin's disease, non-Hodgkin's lymphoma, and myelodysplastic and myeloproliferative syndromes; or 2) non-hematologic malignancies: multiple myeloma, neuroblastoma, and rhabdomyosarcoma.27
In recent times, the possible relationship between RDD and IgG4-related sclerosing disease has been investigated, since both entities share histopathological features such as lymphoplasmacytic infiltrate and stromal fibrosis.18 The pathogenesis of RDD is not clear. There is evidence that it may be associated with an abnormal autoimmune response. The majority of cases of this entity showed abundant plasma cells and sclerosis. Several case reports and recently 2 series of a limited number of cases have demonstrated that plasma cells are expressed in extranodal RDD.
Zhang et al. observed that a significant part of the RDD presents characteristics similar to IgG4-RD, which indicates that there could be an overlap between the two entities. In this study, the number of TREG cells was comparable between nodal RDD and controls, whereas extranodal RDD showed a significantly higher number of TREG cells than controls. This would demonstrate that a subgroup of RDDs has characteristics of IgG4-RD, which is indicative of an overlap between certain aspects of these two entities.28
In the case of the patient reported in this paper, immunohistochemistry of the lymph node, renal, and salivary tissues was performed, in which a high proportion of plasma cells were positive for IgG4 cells. Chen and Lee, meanwhile, observed the presence of stromal fibrosis with abundant plasma cells, which suggests IgG4-RD as a differential diagnosis. In the extranodal involvement, various degrees of stromal fibrosis are observed.29 The marker of this entity is intrasinusal proliferation of S100 histiocytes, which demonstrates emperipolesis and inflammatory cells, as in the case of the patient reported here.
RDD shares histological features with IgG4-RD, namely:
- •
Abundant inflammatory cells.
- •
Various degrees of stromal fibrosis that in some occasions, as in cases of extra-nodal compromise, blur the distinction between these two entities.
RDD disease is part of the differential diagnosis of IgG4-RD, given the histopathological overlap, but the actual relationship between them is unknown. A wide range of therapeutic strategies are currently available. In the asymptomatic patient, observation and control or surgical resection may be indicated if there is a risk of compression of a vital organ. Between 20% and 50% of patients with cutaneous/nodal disease may have spontaneous remissions, which is applicable in cases of uncomplicated cutaneous disease/lymphadenopathy and in those with asymptomatic disease in other sites.
Some cases in which the function of an organ is severely or persistently threatened may require treatment with surgery, steroids (the dose and duration are not defined), chemotherapy (reserved for relapsed or refractory cases, sometimes is used as initial therapy when there is a life-threatening or disseminated disease) or radiotherapy (of modest efficacy in this disease, but it may be beneficial in a disease with refractory soft tissue involvement, orbital commitment, or bone disease with visual impairment or resistant airway obstruction). In 1990, Foucar et al. described 17/238 (7%) patients who died as a result of direct complications of the disease, infections or amyloidosis. Meanwhile, Pulsoni et al. described in 2002 that 10/80 (12%) died as a result of the disease. Patients with multifocal and extranodal disease, particularly those with disease of the lower respiratory tract, liver, and kidney, have a poor prognosis.
Patients with RDD require clinical examinations and laboratory tests every 3–6 months during the first 2 years after complete remission.30
Ethical considerationsThe authors have obtained the informed consent from the patients and/or subjects referred to in the article. This document is in the possession of the corresponding author.
Conflict of interestThe authors declare that they have no conflict of interest.
Please cite this article as: Sager L, Reibaldi A, Calvo R, Ortiz A, Roverano S, Jauk F, et al. Adenopatías cervicales en reumatología: un dilema diagnóstico. Rev Colomb Reumatol. 2022;29:205–213.
