



Bone marrow fibrosis is a rare disorder that can be associated with autoimmune diseases such as systemic lupus erythematosus (SLE), and can be confused with the manifestations of that disease. The case is presented on a patient with autoimmune myelofibrosis in the context of SLE.
La fibrosis de la medula ósea es una patología rara que se puede ver asociada con enfermedades autoinmunes como el lupus eritematoso sistémico (LES) y que puede llegar a ser confundida con las manifestaciones propias de la enfermedad. Se presenta el caso de una paciente con mielofibrosis autoinmune en el contexto de LES.
Bone marrow fibrosis (BMF) is a rare condition associated with different conditions such as neoplastic, infectious, or autoimmune diseases.1 Within the spectrum of autoimmune disorders, it is possible to find myelofibrosis (MF), which is related to systemic lupus erythematosus (SLE), rheumatoid arthritis, antiphospholipid syndrome, autoimmune hemolytic anemia, among others. Although SLE is a disease that frequently has hematological involvement, the association with MF is not common.2 A case of a patient with autoimmune MF secondary to SLE is presented.
Case descriptionA 33-year-old female is referred from the renal unit after an 8-day history of objective fever (38–39 °C), asthenia and adynamia, myalgia, and arthralgia that increased during dialysis. The patient was diagnosed with systemic lupus erythematosus (SLE) at age 17, with cutaneous involvement, and 6 years ago with class V lupus nephritis in renal replacement therapy, along with hematological affection (chronic pancytopenia). She also has a history of arterial hypertension and sulfa allergy, without relevant surgical, toxic, or gynecologic background. Her current medications include prednisone 20 mg/day, chloroquine 250 mg/day, metoprolol 50 mg/day, and calcium plus vitamin D.
Upon physical examination, she has a temperature of 38.6 °C, heart rate of 109/min, blood pressure of 110/70 mm Hg, respiratory rate of 18 per minute, oxygen saturation of 93% with room air, mucocutaneous pallor, and altered consciousness due to drowsiness with episodes of agitation. Additionally, grade III splenomegaly on abdominal palpation was detected. The rest of the physical examination reveals an absence of abnormalities.
Lab tests (Table 1) report pancytopenia, elevated total bilirubin at the expense of direct bilirubin, positive direct Coombs test, a moderate elevation of transaminases, normal complement levels, and negative anti-dsDNA. An abdominal ultrasound reports massive splenomegaly (21 cm).
Lab tests during hospitalization.
| Lab test | Value | Lab test | Value |
|---|---|---|---|
| Leukocytes | 0.7 (103/µl) | Total bilirubin | 4.3 mg/dl |
| Neutrophils | 400 (abs/mm3) | Direct bilirubin | 4.1 mg/dl |
| Lymphocytes | 100 (abs/mm3) | Corrected reticulocyte count | 0.3% |
| Monocytes | 200 (abs/mm3) | Direct Coombs test | +++ |
| Hemoglobin | 5.5 g/dl | C3 Complement | 94 (88−165) |
| Hematocrit | 18.2% | C4 Complement | 18.3 (14−44) |
| Platelets | 26 (109/µl) | Anti-DNA | Negative |
| Aspartate aminotransferase | 207 U/L | Alanine aminotransferase | 86 U/L |
The clinical approach was based on the diagnosis of febrile neutropenia, so broad-spectrum antibiotic coverage with vancomycin and piperacillin-tazobactam was started for one week, in addition to requesting tests for systemic infection, especially of the central nervous system; such studies were initially negative. Subsequently, extended-spectrum beta-lactamase (ESBL) Klebsiella pneumoniae bacteremia was documented and treated for 14 days with meropenem with negative control cultures.
Due to the persistence of fever in a patient with pancytopenia, immunosuppression, and extensive antibiotic regimen spectrum, anidulafungin coverage due to suspicion of fungemia was initiated, with final negative cultures. In the presence of massive splenomegaly and abnormal liver tests, studies were expanded; chronic liver disease was ruled out, as well as portal hypertension.
Finally, it was considered that the aforementioned alterations were secondary to lupus activity with neuropsychiatric, hematological, and gastrointestinal manifestations (lupoid hepatitis). Intravenous methylprednisolone pulse therapy (250 mg/day for 3 doses) was started, with a resolution of neuropsychiatric manifestations and normalization of hepatic biochemical tests, yet with the persistence of cytopenias, reason why a bone marrow biopsy was performed, which was not indicative of malignancy. The sample was not adequate, since it was hemodiluted, which made it necessary to perform a new one. It was decided to start rituximab due to haematological lupus activity in the context of persistence of severe pancytopenia (Table 2); however, the patient presented multiple infectious episodes during hospitalization that delayed this process.
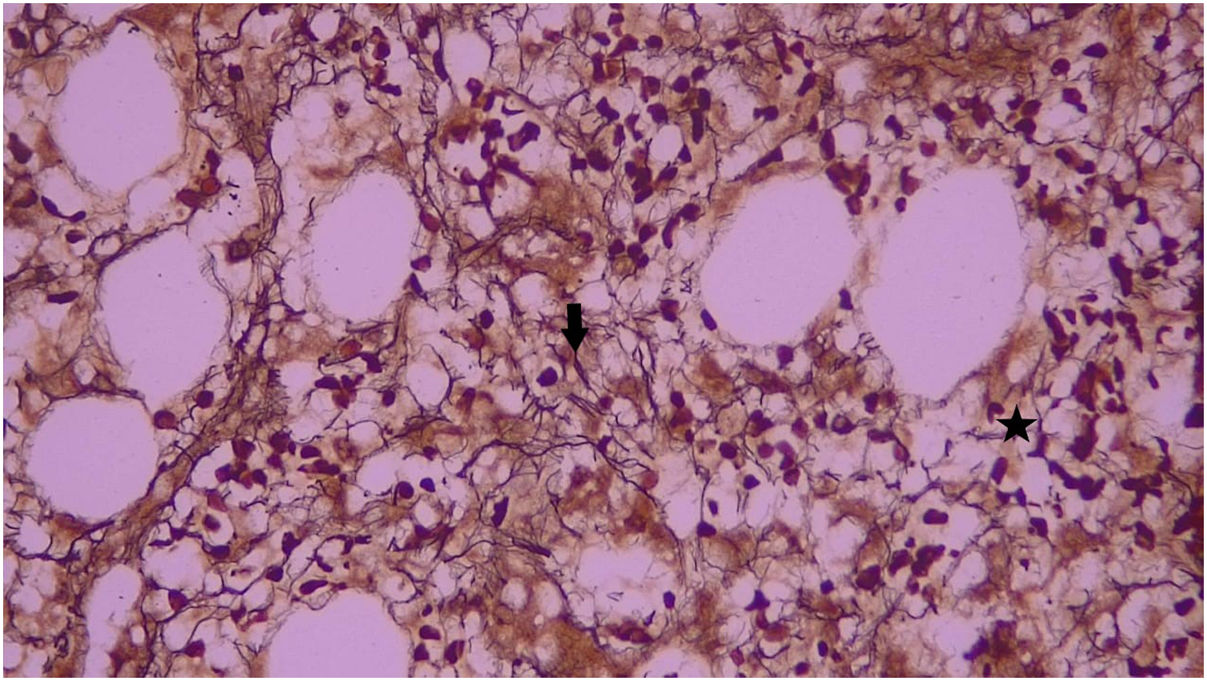
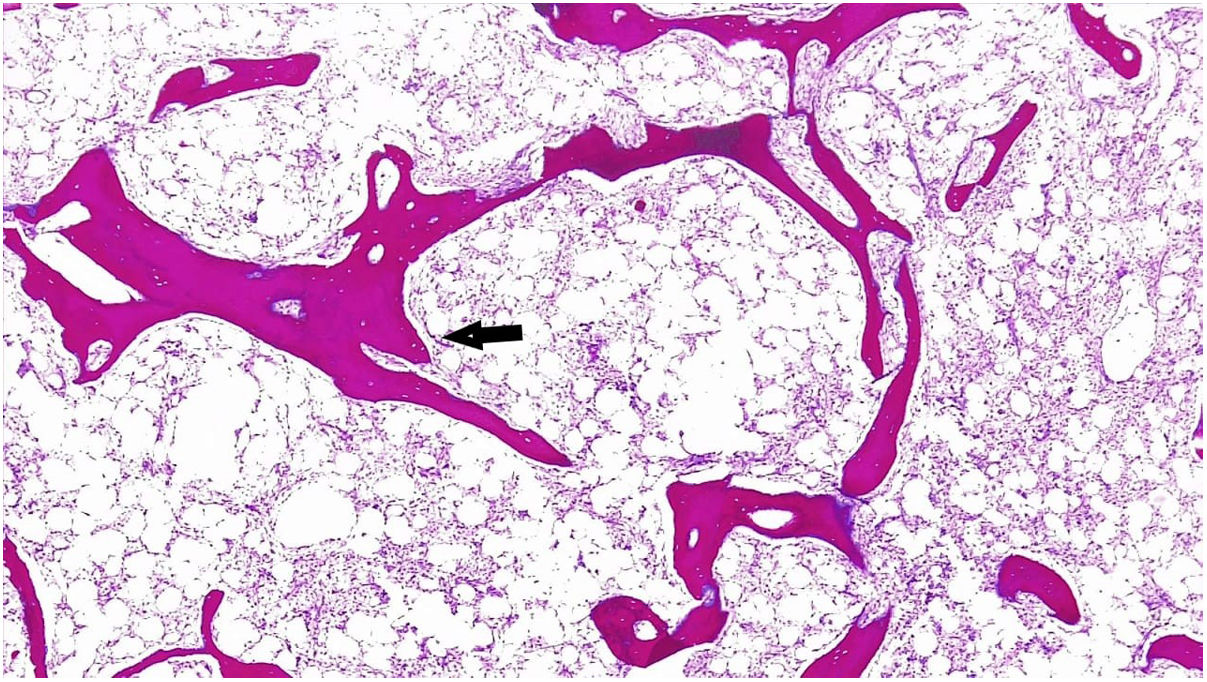
The report of the second bone marrow biopsy revealed subcortical representation with areas of emptying and variable cellularity (5–30%), a myeloid/erythroid ratio of 2:1, erythroid line with adequate maturation, and morphology with a tendency to formation of small aggregates. The granulocytic line was diminished, with maturation arrest, and megakaryocytes up to 5 per high-power field were found, some of them with slight morphological atypia due to nuclear hyperchromatism. The special reticular stain demonstrated fine reticular fibers with frequent intersections (grade I of Thiele classification). No deposits of mature collagen are observed. The above findings correspond to bone marrow with variable cellularity, decreased granulocytic and erythroid lines, and morphological changes suggestive of grade I-II Thiele myelofibrosis (MF) (Figs. 1 and 2).
 and other areas with dense intersections (arrow) 40×. It corresponds to grade I-II reticulin myelofibrosis of the Thiele classification.")
 4×.")
Investigations were expanded to rule out primary MF and other etiologies of massive splenomegaly, with negative studies for JAK2 mutation and Gaucher disease. A diagnosis of SLE-associated MF was proposed. The first dose of anti-CD20 was administered, but the patient again presented infectious complications secondary to severe pancytopenia, such as a new episode of ESBL Klebsiella pneumoniae bacteremia, treated with ertapenem for 10 days, in addition to transfusional support. During her stay, the patient did not require transfer to the intensive care unit; nonetheless, she later presented a progressive clinical deterioration that led to death.
DiscussionBone marrow fibrosis (BMF) is associated with hematologic and non-hematologic conditions. Regarding its pathogenesis, the interaction of polymorphonuclear and stromal cells through the intervention of proinflammatory cytokines such as TGF-β leads to this disorder.3
Primary MF, which is the most common presentation, is secondary to a clonal disorder of hematopoietic cells. Among the secondary causes, autoimmune MF was described, as mainly related to SLE and, to a lesser extent, to Sjögren's syndrome, autoimmune hepatitis, rheumatoid arthritis, antiphospholipid syndrome, and autoimmune hemolytic anemia, among others.
There are differences between primary and autoimmune MF, such as a higher degree of fibrosis, presence of hypercellularity, and reactive lymphoid aggregates in autoimmune etiology, especially in those over 60 years of age, attributable to the underlying disease. It is noteworthy that this disease can also be secondary to infections like tuberculosis, human immunodeficiency virus, and visceral leishmaniasis.3
In the case of SLE, haematological involvement is common, generally due to peripheral destruction, deficiency, infection, or secondary to medications. Direct affection of the bone marrow is rare, and even more the presence of MF, which is more common in middle-aged women with an established diagnosis.4–6 The incidence of SLE in patients with autoimmune MF ranges around 72–76%, being the most frequently associated connective tissue disease; however, it may be underestimated or underreported, taking into account that the manifestations can be easily related to autoimmune disease.2,4 It should be suspected in patients with pancytopenia, as occurred with the current case, or in the presence of single cytopenias such as refractory anemia.
Among the characteristic features in the bone marrow analysis, T and B lymphocytes aggregates in the absence of morphological and clinical characteristics of myeloproliferative or lymphoproliferative diseases are found. Similarly, to confirm the diagnosis of autoimmune MF, the presence of reticular fibrosis in the bone marrow must be documented and other causes that explain these findings must be ruled out.1,7 In this case, the presence of changes related to fibrosis in the bone marrow is evident, with extension studies that exclude other aetiologies.8
Only one case series found a high prevalence of MF in subjects with SLE and cytopenias.Üsküdar Cansu et al. included 41 individuals with these characteristics who underwent bone marrow biopsy, of whom 29 (69%) were positive for MF.9 Although hematological abnormalities in SLE tend to improve with the use of glucocorticoids and immunosuppressants, when MF is associated, the rate and response time are low (response time of patients with SLE, cytopenias, and presence or absence of MF were 3.3±3.1 months vs. 1.7±1.2 months, respectively p = 0.091).9,10
In the current case, the patient received a dose of rituximab without obtaining the expected results. Initially, it was considered that this could be related to the degree of MF. However, there are case reports with greater severity with an adequate response, so it is possible that the outcome is related, in addition to changes in the bone marrow, to the infectious processes and the multiple subsequent complications.11
As for splenomegaly in the SLE scenario, which occurs with a prevalence of 9–46 %, as indeed occurs in this case, it is commonly reported in autoimmune MF.2,12 It should be considered as a parameter of disease activity, responding adequately to corticosteroids. Considering its etiology, it is not clear, but it has been attributed to the obstruction of the flow due to the presence of thrombi. Massive splenomegaly (>18 cm) is uncommon, thus, in this context, infectious (especially leishmaniasis and malaria), neoplastic (lymphomas) and, deposit (Gaucher) diseases must be considered,12 as was done in this report.
In conclusion, autoimmune MF is a condition that should be considered and, therefore, ruled out in patients with pancytopenia or hematological involvement in the setting of connective tissue diseases, especially SLE, in which the greatest association has been described.
Informed consentThe authors declare that signed informed consents were obtained from the patients or their parents, as well as the approval of the ethics committee of the institution.
FinancingFor the preparation of this article, each co-author's resources were used.
Conflict of interestsThe authors declare that they have no conflict of interest.
Please cite this article as: Guavita D, Cajamarca J, Mendez J, Moreno L, Arredondo AM, Hernán Cubides H, et al. Mielofibrosis autoinmune y lupus eritematoso sistémico: reporte de un caso. Rev Colomb Reumatol. 2022;29:214–217.
