



Systemic sclerosis is an autoimmune and multisystemic disease characterized by vascular involvement and fibrosis that mainly affects the skin and internal organs; its morbidity and mortality are the highest of rheumatic diseases.
ObjectiveTo determine the sociodemographic, clinical, and paraclinical characteristics of adult patients with systemic sclerosis in a reference center in rheumatology in northwestern Colombia, between 2006 and 2016.
MethodsA retrospective descriptive study was conducted. Information on sociodemographic, clinical, and paraclinical variables was collected from the review of medical records. The data were processed with the IBM SPSS 22 statistical package. The qualitative variables were expressed in absolute and relative frequencies and the quantitative variables in mean and standard deviation or median and interquartile range, according to their distribution.
Results44 patients were included, predominantly women (90.9%), with an average age of 59 years; the most common variety was the limited (61.3%). One hundred percent of the subjects presented Raynaud's phenomenon and the most common compromised systems were: cutaneous (100%), musculoskeletal (59.1%), and gastrointestinal (50%). 95.5% of the patients fulfilled the diagnostic criteria ACR / EULAR 2013. Regarding the immunological profile, 72.7% of the individuals had positive antinuclear antibodies, of which 53.1% had a centromeric pattern.
ConclusionsSociodemographic, clinical and similar characteristics were found in this cohort, according to what was published in the literature. The most frequent extracutaneous systems involved were: musculoskeletal, gastrointestinal, and pulmonary, which cause a large part of the associated morbidity, as well as a decrease in quality of life.
La esclerosis sistémica es una enfermedad autoinmune y multisistémica caracterizada por vasculopatía y fibrosis que afecta principalmente la piel y órganos internos; su morbilidad y mortalidad son las más altas de las enfermedades reumatológicas.
ObjetivoDeterminar las características sociodemográficas, clínicas y paraclínicas de pacientes adultos con esclerosis sistémica en un centro de referencia en reumatología del noroccidente de Colombia, entre 2006 y 2016.
MétodosEstudio descriptivo retrospectivo. La información sobre las variables sociodemográficas, clínicas y paraclínicas se recogió de la revisión de registros médicos. Los datos se procesaron con el paquete estadístico IBM SPSS 22. Las variables cualitativas se expresaron en frecuencias absolutas y relativas y las cuantitativas en media y desviación estándar o mediana y rango intercuartílico, según su distribución.
ResultadosSe incluyeron 44 pacientes, predominantemente mujeres (90,9%), con una edad promedio de 59 años; la variedad más común fue la limitada (61,3%). El 100% de los sujetos presentaron el fenómeno de Raynaud y los sistemas comprometidos más comunes fueron: cutáneo (100%), musculoesquelético (59,1%) y gastrointestinal (50%). El 95,5% de los pacientes cumplieron con los criterios diagnósticos ACR / EULAR 2013. Con respecto al perfil inmunológico, el 72,7% de los individuos tenía anticuerpos antinucleares positivos, de los cuales un 53,1% tenía un patrón centromérico.
ConclusionesSe encontraron características sociodemográficas, clínicas y características similares en esta cohorte, de acuerdo a lo publicado en la literatura. Los sistemas extracutáneos más frecuentes involucrados fueron: musculoesquelético, gastrointestinal y pulmonar, que causan una gran parte de la morbilidad asociada, así como una disminución en la calidad de vida.
Systemic sclerosis (SS) is a chronic autoimmune disease characterized by varying degrees of vasculopathy and fibrosis, affecting different organs such as the skin, gastrointestinal tract, and cardiorespiratory system.1,2 Although its cause and pathophysiology are unknown, it is known that at least three phenomena occur: endothelial dysfunction, clinically evident as Raynaud's phenomenon (present in 95% of patients),3 activation of fibroblasts with subsequent collagen deposition and cutaneous fibrosis, and production of autoantibodies.4
Its incidence and prevalence vary depending on the geographical location, being between 0.6 and 23 per million inhabitants per year and 7–489 cases per million inhabitants, respectively.5–7 Its male–female ratio is 6:1 and the age group most frequently affected is between 45 and 64 years.8,9 Clinically, this disease can be subclassified into 3 groups: the limited form – characterized by the predominance of cutaneous distal involvement and pulmonary arterial hypertension (PAH) as a complication –,10 the diffuse form with extensive skin involvement and interstitial lung disease (ILD)1,11 and the sine scleroderma form, in which the organic compromise predominates affecting less than 5% of patients.12 Despite its low prevalence, it has the highest rate of mortality and impact on the quality of life between rheumatic diseases, given primarily for its pulmonary complications: PAH and ILD.1,2
As far as we know, in Latin America, few data are available, coming from Argentina, Dominican Republic, and Brazil.4,8 In Colombia, there is one study about the prevalence of the disease by Fernandez-Avila et al., in which after reviewing the diagnostic codes they calculated a prevalence of 23.7/100,000, relatively similar to other countries in the region.13 Some studies have reported the clinical and paraclinical characteristics, as well as complications and mortality. Among the most influential, there are two articles from the Universidad Nacional de Colombia and Universidad CES; the latter emphasizing the pulmonary complications of the disease.14,15 On the other hand, after an exhaustive review of the literature, no information was found from local or regional cohorts that have used the current classification criteria for the disease.16
For these reasons, this work aimed to determine what are the sociodemographic, clinical, paraclinical and therapeutic features of patients with systemic sclerosis in a cohort of a rheumatology reference center in northwestern Colombia, in light of the current classification criteria.
Materials and methodsStudy design and populationA retrospective descriptive study was conducted among adult patients diagnosed with SS, in a reference center that attended both inpatient and outpatient subjects between 2006 and 2017. Patients over 18 years, with a diagnosis of SS, were included based on the 2013 ACR/EULAR classification criteria16 or classified by their treating rheumatologist. Subjects with other autoimmune diseases that could generate confounding biases (rheumatoid arthritis, vasculitis, lupus, antiphospholipid syndrome, ankylosing spondylitis, among others) were excluded.
Collection processSociodemographic (sex, age, place of residence, comorbidities), clinical (SS type, cutaneous and visceral organic involvement), and paraclinical variables related to the disease (images, antibodies, capillaroscopy) were evaluated. A system affection was considered present where there was a specific test that demonstrates it or if in the clinical chart there was specific mention of such compromise by the treating physician.
Prior endorsement of the Research Ethics Committee, the information was collected by reviewing the physical and electronic medical records that had the diagnostic codes of SS in the ICD 10: M340, M341, M348, and M349. The data obtained were entered into the MAGPI™, an electronic platform that allows creating online formularies for any specific purpose. After designing a formulary specifically to collect the desired variables, we conducted a pilot test with the first 20 patients, to make adjustments in the collection instrument. In case of doubts, these were resolved in a joint review by the rheumatology researchers. Once the information was collected, it was exported to Microsoft Excel 2016 spreadsheet, where a categorization of the quantitative variables that required it and coding of the qualitative variables were performed; and the consistency of the data was evaluated before their analysis.
Statistical analysisThe qualitative variables were expressed in absolute and relative frequencies and the quantitative ones in mean and standard deviation or median and interquartile range (IQR), according to their distribution. The analysis was carried out in the IBM SPSS 22 statistical package.
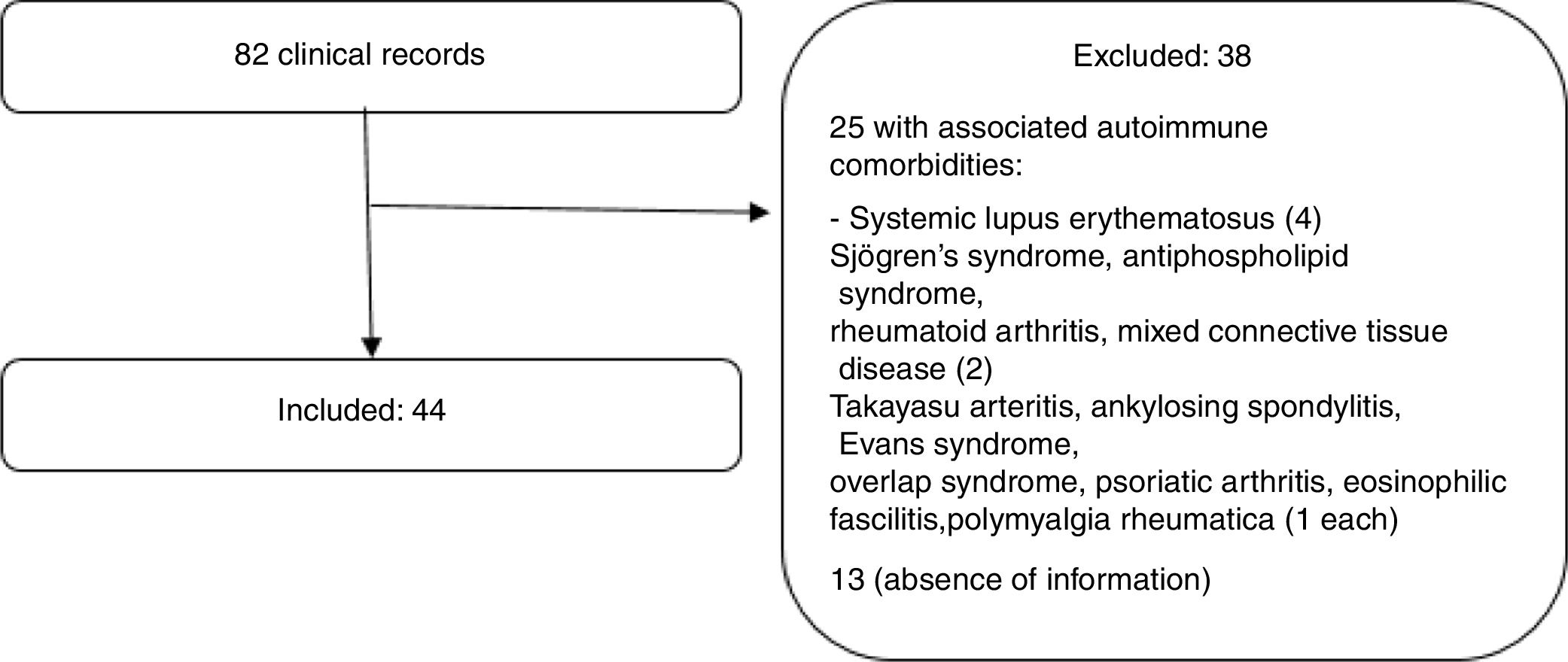
ResultsProcess of population selectionOf 105 clinical records that had SS diagnosis codes, 23 were not included in the analysis due to misclassification. Finally, 82 histories were analyzed; 13 were excluded because of lack of information (a single visit to the rheumatologist without full clinical data); and 25 for associated autoimmune disease. In total, 44 patients met the eligibility criteria. Fig. 1 depicts the selection process of the subjects.
Sociodemographic features
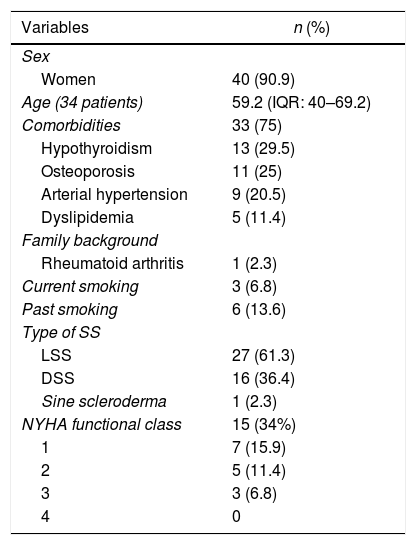
Of a total of 44 patients included in the analysis, 90.9% were women, with a median age of 59 years, and the most common area of residence was urban in 84%. Thirty-three individuals (75%) had, at least, one comorbidity, being the most frequent: hypothyroidism, osteoporosis, and arterial hypertension. Other associated diseases were neoplasms (n=4; 9.1%), chronic kidney disease (n=3; 6.8%), diabetes mellitus (n=2; 4.5%), fibromyalgia (n=2; 4.5%), psychiatric disorders (n=2; 4.5%), and coronary artery disease, heart failure, venous insufficiency, migraine, and osteoarthritis (one case each; 2.3%). One patient had a report of use of psychoactive substances, and one subject had a family history of autoimmune disease (rheumatoid arthritis). The most frequent subtype was limited systemic sclerosis (LSS) (n=27; 61.3%), followed by diffuse (DSS) (n=16; 36.4%) and sine scleroderma (n=1; 2.3%). The detailed information on sociodemographic characteristics is illustrated in Table 1.
Sociodemographic characteristics of a cohort of patients with systemic sclerosis of northwestern Colombia.
| Variables | n (%) |
|---|---|
| Sex | |
| Women | 40 (90.9) |
| Age (34 patients) | 59.2 (IQR: 40–69.2) |
| Comorbidities | 33 (75) |
| Hypothyroidism | 13 (29.5) |
| Osteoporosis | 11 (25) |
| Arterial hypertension | 9 (20.5) |
| Dyslipidemia | 5 (11.4) |
| Family background | |
| Rheumatoid arthritis | 1 (2.3) |
| Current smoking | 3 (6.8) |
| Past smoking | 6 (13.6) |
| Type of SS | |
| LSS | 27 (61.3) |
| DSS | 16 (36.4) |
| Sine scleroderma | 1 (2.3) |
| NYHA functional class | 15 (34%) |
| 1 | 7 (15.9) |
| 2 | 5 (11.4) |
| 3 | 3 (6.8) |
| 4 | 0 |
IQR: interquartile range; SS: systemic sclerosis; LSS: limited systemic sclerosis; DSS: diffuse systemic sclerosis; NYHA: New York Heart Association.
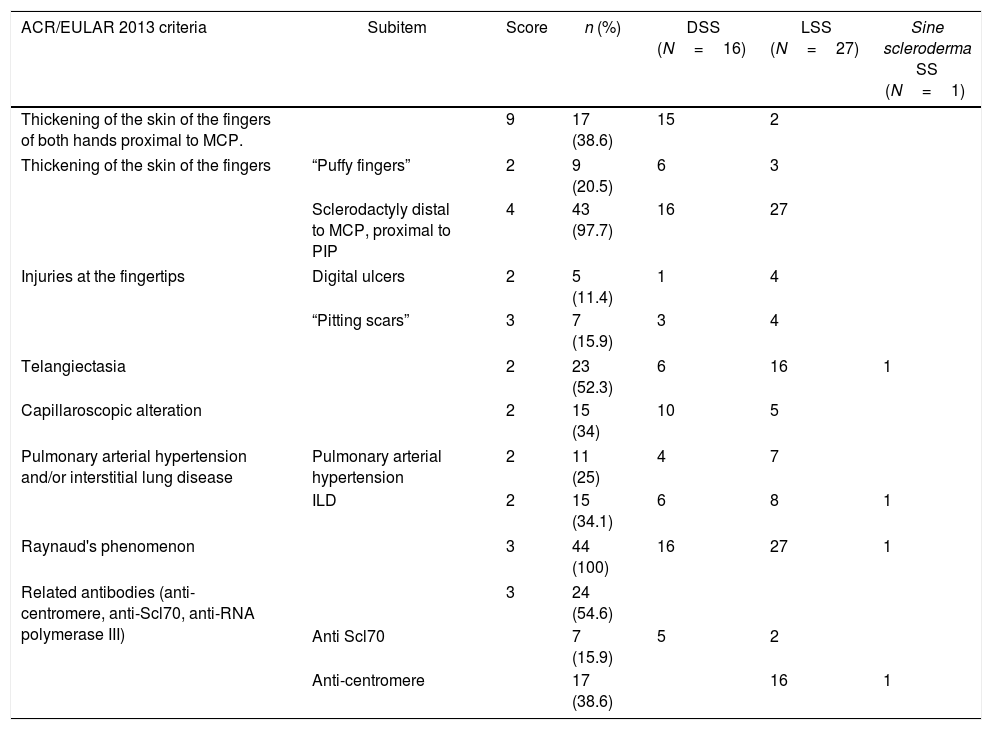
95.5% of the subjects included had a score of 9 in 2013 ACR/EULAR criteria; of these, the most frequent were: Raynaud's phenomenon (n=44; 100%) and sclerodactyly (n=43; 97.7%). Seventeen patients (38.6%) met the criterium of thickening of the skin of the fingers of both hands proximal to metacarpophalangeal joints (MCP), which is sufficient for the diagnosis of the disease. In two individuals (4.5%) a score lower than 9 was obtained; in these cases, the diagnosis was based on the thickening of the skin of the fingers, digital injuries, positivity of the anti-centromere antibodies, and visceral organ involvement (interstitial pulmonary and esophageal). The details of the classification criteria are shown in Table 2.
ACR/EULAR 2013 classification criteria in a cohort of patients with systemic sclerosis in northwestern Colombia.
| ACR/EULAR 2013 criteria | Subitem | Score | n (%) | DSS (N=16) | LSS (N=27) | Sine scleroderma SS (N=1) |
|---|---|---|---|---|---|---|
| Thickening of the skin of the fingers of both hands proximal to MCP. | 9 | 17 (38.6) | 15 | 2 | ||
| Thickening of the skin of the fingers | “Puffy fingers” | 2 | 9 (20.5) | 6 | 3 | |
| Sclerodactyly distal to MCP, proximal to PIP | 4 | 43 (97.7) | 16 | 27 | ||
| Injuries at the fingertips | Digital ulcers | 2 | 5 (11.4) | 1 | 4 | |
| “Pitting scars” | 3 | 7 (15.9) | 3 | 4 | ||
| Telangiectasia | 2 | 23 (52.3) | 6 | 16 | 1 | |
| Capillaroscopic alteration | 2 | 15 (34) | 10 | 5 | ||
| Pulmonary arterial hypertension and/or interstitial lung disease | Pulmonary arterial hypertension | 2 | 11 (25) | 4 | 7 | |
| ILD | 2 | 15 (34.1) | 6 | 8 | 1 | |
| Raynaud's phenomenon | 3 | 44 (100) | 16 | 27 | 1 | |
| Related antibodies (anti-centromere, anti-Scl70, anti-RNA polymerase III) | 3 | 24 (54.6) | ||||
| Anti Scl70 | 7 (15.9) | 5 | 2 | |||
| Anti-centromere | 17 (38.6) | 16 | 1 | |||
DSS: diffuse systemic sclerosis; LSS: limited systemic sclerosis; SS: systemic sclerosis; MCP: metacarpophalangeal joints; PIP: proximal interphalangeal joints; ILD: interstitial lung disease.
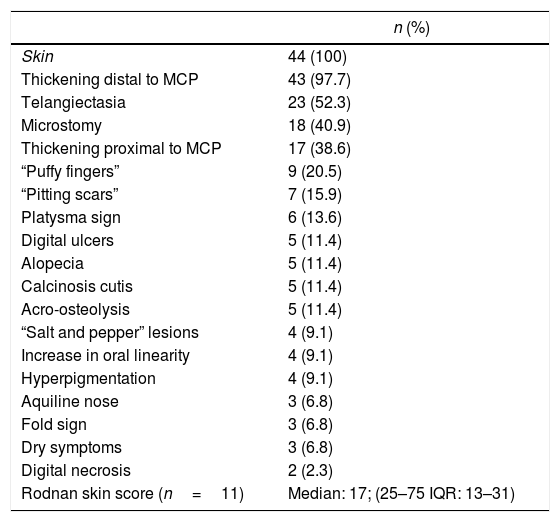
Of the patients with Raynaud's phenomenon, in 15 (34%) the vascular phases were described: pallor (n=8; 53.3%), cyanosis (n=7; 46.6%), and erythema (n=4; 26.6%). The first non-Raynaud symptom was reported in 32 individuals (72.7%), being the most frequent: arthralgia (n=10; 31.2%), followed by skin sclerosis (n=8; 18.2%), and “puffy fingers” (n=4.9; 1%). The Rodnan skin score was available in 11 patients, with a median of 17 points and a mean of 20.27. The other cutaneous findings are summarized in Table 3.
Cutaneous manifestations of a cohort of patients with systemic sclerosis of northwestern Colombia.
| n (%) | |
|---|---|
| Skin | 44 (100) |
| Thickening distal to MCP | 43 (97.7) |
| Telangiectasia | 23 (52.3) |
| Microstomy | 18 (40.9) |
| Thickening proximal to MCP | 17 (38.6) |
| “Puffy fingers” | 9 (20.5) |
| “Pitting scars” | 7 (15.9) |
| Platysma sign | 6 (13.6) |
| Digital ulcers | 5 (11.4) |
| Alopecia | 5 (11.4) |
| Calcinosis cutis | 5 (11.4) |
| Acro-osteolysis | 5 (11.4) |
| “Salt and pepper” lesions | 4 (9.1) |
| Increase in oral linearity | 4 (9.1) |
| Hyperpigmentation | 4 (9.1) |
| Aquiline nose | 3 (6.8) |
| Fold sign | 3 (6.8) |
| Dry symptoms | 3 (6.8) |
| Digital necrosis | 2 (2.3) |
| Rodnan skin score (n=11) | Median: 17; (25–75 IQR: 13–31) |
MCP: metacarpophalangeal joints; IQR: interquartile range.
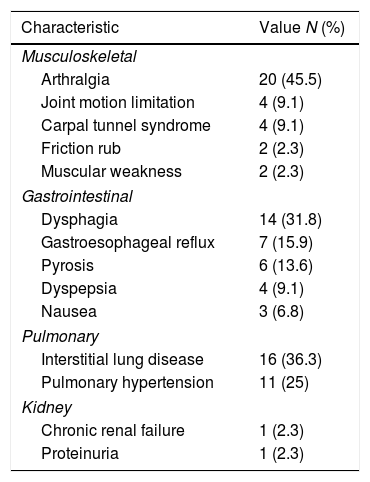
The most frequent extra-cutaneous involvement was musculoskeletal (n=26; 59.1%), followed by gastrointestinal (n=22; 50%), and pulmonary (n=20; 45.5%) which are depicted in Table 4. Regarding interstitial lung disease (ILD), even though we had access to radiology reports, they did not specify which type of ILD was present at the moment; therefore, they could only be classified globally as ILD. The same situation occurred with spirometry, in which the results had to be dichotomized into normal or restrictive; none of the subjects had an obstructive pattern.
Extra cutaneous affection in a cohort of patients with systemic sclerosis in northwestern Colombia.
| Characteristic | Value N (%) |
|---|---|
| Musculoskeletal | |
| Arthralgia | 20 (45.5) |
| Joint motion limitation | 4 (9.1) |
| Carpal tunnel syndrome | 4 (9.1) |
| Friction rub | 2 (2.3) |
| Muscular weakness | 2 (2.3) |
| Gastrointestinal | |
| Dysphagia | 14 (31.8) |
| Gastroesophageal reflux | 7 (15.9) |
| Pyrosis | 6 (13.6) |
| Dyspepsia | 4 (9.1) |
| Nausea | 3 (6.8) |
| Pulmonary | |
| Interstitial lung disease | 16 (36.3) |
| Pulmonary hypertension | 11 (25) |
| Kidney | |
| Chronic renal failure | 1 (2.3) |
| Proteinuria | 1 (2.3) |
In this cohort, 32 (72.7%) patients had a report of antinuclear antibodies (HEP2-IFI) in the medical chart, all of them were positive, predominantly with centromere pattern (n=17; 53.1%), followed by homogeneous and nucleolar (both present in five subjects – 15.6%); in all patients but one the dilution of HEP2-IFI was 1/160 or superior. Anti-Scl70 antibodies were reported in 20 subjects (45.4%), being positive in seven (35%). Anti-extractable nuclear antigens (ENAS) were requested in 23 cases (52.3%), being positive in two (8.7%), specifically anti-Ro antibodies.
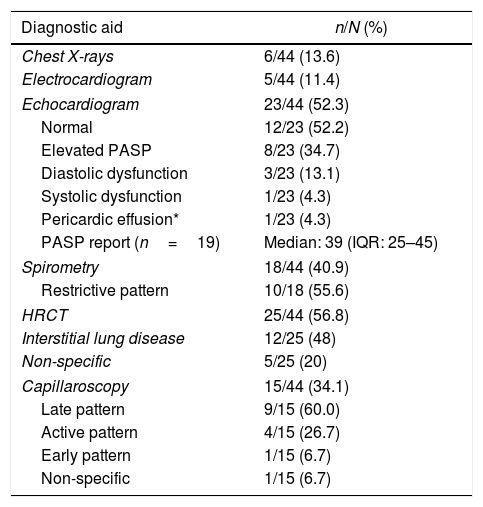
The most requested diagnostic aids were: echocardiogram (n=23; 52.3%), thorax high-resolution computed tomography (HRCT) (n=25; 56.8%), and chest X-rays (n=6; 13.6%). High values of the pulmonary artery systolic pressure were reported on echocardiography in 19 subjects (Table 5).
Diagnostic aids in a cohort of patients with systemic sclerosis in northwestern Colombia.
| Diagnostic aid | n/N (%) |
|---|---|
| Chest X-rays | 6/44 (13.6) |
| Electrocardiogram | 5/44 (11.4) |
| Echocardiogram | 23/44 (52.3) |
| Normal | 12/23 (52.2) |
| Elevated PASP | 8/23 (34.7) |
| Diastolic dysfunction | 3/23 (13.1) |
| Systolic dysfunction | 1/23 (4.3) |
| Pericardic effusion* | 1/23 (4.3) |
| PASP report (n=19) | Median: 39 (IQR: 25–45) |
| Spirometry | 18/44 (40.9) |
| Restrictive pattern | 10/18 (55.6) |
| HRCT | 25/44 (56.8) |
| Interstitial lung disease | 12/25 (48) |
| Non-specific | 5/25 (20) |
| Capillaroscopy | 15/44 (34.1) |
| Late pattern | 9/15 (60.0) |
| Active pattern | 4/15 (26.7) |
| Early pattern | 1/15 (6.7) |
| Non-specific | 1/15 (6.7) |
PASP: pulmonary artery systolic pressure; HRCT: thorax high-resolution computed tomography; IQR: interquartile range.
Regarding capillaroscopy, it was reported in 15 patients (34.1%); a SS pattern was found in 14 subjects (93.3%), being the late pattern the predominant one.
TreatmentAt the time of admission to the cohort, 30 subjects (68.2%) had received prior treatment, being calcium-channel antagonists (n=19; 43.3%), proton pump inhibitors (n=13; 29.5%), selective serotonin reuptake inhibitors (n=9; 20.5%), and prednisone (n=8; 18.2%) the most used. In the first visit, treatment was started in 24 subjects (54%), more often with calcium-channel antagonists (n=15; 34.1%), selective serotonin reuptake inhibitors (n=11; 25%), acetylsalicylic acid, and prednisolone (n=8; 18.2%, each one). In 14 individuals (32%) immunomodulation therapy was initiated, being methotrexate and cyclophosphamide the most prescribed agents (n=12; 27-3% and n=11; 25%, respectively). Of the patients of the cohort, nine (20.5%) required hospitalization in the first consultation, being the most common cause disease activity in eight cases (88.9%).
DiscussionThe most striking finding of this study is that 95.5% of the patients of the cohort met the current classification criteria for systemic sclerosis, information not reported, as far as we know, in Colombia or Latin America. The subjects included in this study presented sociodemographic characteristics like those described in other national and international reports, such as the predominance of female sex and a higher prevalence between 40 and 64 years.7,8,17,18
In the literature, there are reports of SS subtypes classification according to the region analyzed; for example, in Latin America predominated the limited type (72%), with variable percentages in Europe (61%) and North America (53%). These data are similar to those obtained in the current cohort. Moreover, the frequency of the sine scleroderma type was similar to that reported in the literature.12,18
Regarding clinical aspects, the frequency of Raynaud's phenomenon was similar to that reported in the literature (94–96%).19 Additionally, it is noteworthy that there was a low incidence of digital ulcers compared with other cohorts such as the reported by Sambova et al. (35%).20 This finding could have several explanations, such as a low threshold of detection by the treating physician which would be unlikely taking into account that digital ulcers are usually symptomatic and we are a reference center with rheumatologists trained in the management of SS. Second, it could be explained by our average temperature of 28°C; this fact could protect against vasculopathic manifestations of the disease.
We highlight that, in a high percentage, the diagnosis of SS in this cohort was established only with the finding on physical examination of thickening of the skin proximal to the metacarpophalangeal joints, an essential contribution of the 2013 classification criteria16 which allows this unique finding to be enough as a diagnostic test and enables timely referral of these subjects.
Another interesting fact of this cohort is that it is possible to make the diagnosis of SS without cutaneous thickening, considering digital findings, visceral organic involvement and autoantibodies, adding each one specific value, achieving the required score for the diagnosis. These findings confirm the adequate diagnostic performance of the SS current classification criteria, with a reported sensitivity of 79.4% in daily clinical practice.21
Regarding the complications, the percentage of interstitial lung involvement is similar to that reported in other cases of SS, especially with the limited type (35–52%).5 One aspect to improve is that a high percentage of echocardiograms with increased systolic pulmonary artery pressure was found and there was no evidence of application of flow charts to decide the relevance of right catheterization; this behavior allows a systematic application of this diagnostic aid and, if the diagnosis of pulmonary arterial hypertension (PAH) is confirmed, specific vasodilator treatment should be initiated, with evidence of reduction of long-term morbidity and mortality.22
We highlight the high frequency of joint involvement as a first non-Raynaud symptom, finding that could be explained because the cohort comes from a rheumatologic center where, probably, patients were referred due to joint affection as a dominant symptom. The frequency of this finding contrasts with the report of Dougherty et al., where they found a lower frequency of joint involvement (24.6%).23
Regarding the results of antinuclear antibodies, the centromere pattern was the most frequent, finding expected for a cohort with a predominance of the limited type. It is striking that the second pattern reported in frequency, with the nucleolar, was homogeneous; this finding could be explained by the presence of anti-Pm/Scl antibodies, which are not routinely requested in daily practice; the frequency of this antibody reported in the literature is 7.2%.24 Additionally, it was striking the presence of anti-Ro antibodies in two subjects, which reinforces the notion that this antibody may be present in other diseases different to Sjögren syndrome; for example, Tao et al. found anti-Ro antibodies in 6/28 subjects in a SS cohort.25
Another interesting finding in this cohort was the high frequency of cyclophosphamide use and requirement of hospitalization in the first consultation, mostly due to high disease activity; this reflects probably a late remission and reflects the need to sensitize the primary care physician and other specialists about the early signs and symptoms of this disease.26 This finding also should underscore the need for early sclerosis cohorts which can have different characteristics and could be missed in our cohort.
Regarding the capillaroscopic findings, it was striking the high frequency of the late pattern in this cohort, showing an advanced process of microvascular damage; the frequency of this pattern was greater than that reported by Ingegnoli et al., which was 31.9%.27 This again reinforces the fact that this is indeed a late disease cohort that probably reflects a specific moment in the natural progression of SS.
As for the strengths of the present survey, we had an extensive data search and collected all the available clinical variables, which allowed us to provide a detailed characterization of some aspects as Raynaud phenomenon, clinical findings, capillaroscopy, and the fulfillment of the 2013 classification criteria, which, as far as we know, it has not been reported previously in national or regional cohorts. Regarding the weakness, we recognized that there were several variables with incomplete data which hinders the capacity of getting all the relevant information. Being the present study based on clinical records, this flaw is not susceptible to improvement but highlights the importance of keeping such records in a tidy manner to improve the data quality, ideally in an ordered coordinated fashion allowing multicentric studies. The second limitation was the fact that we are a reference center, as such our results may not represent the complete spectrum of the disease. To minimize this issue, we used all the relevant codes for SS. Also, SS is a complex disease and is usually managed in reference centers; so, it is unlikely that some less complex centers could have a significant amount of patients. For these reasons, our patients were more representative of more advanced disease and there are international early SS cohorts showing that detecting these patients should be a priority, both in research and clinical settings, to provide expedited care to this subpopulation.
ConclusionsIn a cohort of patients with systemic sclerosis from a reference center in northwestern Colombia, the subjects fulfilled, in a high percentage, the current classification criteria of the disease, especially cutaneous thickening proximal to the metacarpophalangeal joints, a unique finding that confirms this entity. It was also striking the high frequency of joint involvement as an extra-cutaneous system, the capillaroscopic evidence of a late pattern and the under-registration or non-application of diagnostic flow diagrams for the detection of pulmonary arterial hypertension.
FundingOwn resources.
Conflict of interestsNone declared.
To Universidad Pontificia Bolivariana and Clinica Universitaria Bolivariana for facilitating the development of this work.
Please cite this article as: Ferreira Morales J, Gutiérrez Tamayo AM, de Olaya EB, Rodríguez Padilla LM, Velásquez-Franco CJ, Mesa Navas MA. Úlceras digitales en esclerosis sistémica. Rev Colomb Reumatol. 2020;27:2–9.
