



Chilblain lupus is a rarely manifested variant of chronic lupus. Its appearance can be sporadic or hereditary associated with an autosomal dominant mutation. The diagnosis is clinical and histopathological. The case is presented of a patient with systemic lupus erythematosus presenting with chilblain lupus and nail involvement, despite the use of antimalarials and immunomodulators.
La perniosis lúpica es una variante del lupus crónico que se manifiesta con poca frecuencia, su aparición puede ser esporádica o hereditaria, asociada con una mutación autosómica dominante, en tanto que su diagnóstico es clínico e histopatológico. Se reporta el caso de una mujer con lupus eritematoso sistémico con manifestación de perniosis lúpica y compromiso ungueal, a pesar del uso de antimaláricos e inmunomoduladores.
Chilblain lupus is a rare form of presentation of cutaneous lupus erythematosus (CLE). The first description was made by Jonathan Hutchinson in 1888, after observing lesions on the hands, feet, and ears, secondary to exposure to low temperatures in patients with systemic lupus erythematosus (SLE).1,2 In the CLE series, chilblain lupus represented 3%–11% of cases, while in long-term follow-up, about 18% met SLE criteria.2
The terms pernio, perniosis, erythema pernio, and chilblain describe skin lesions that appear after exposure to cold without an associated underlying disease and correspond to the Anglo-Saxon term: “chilblain”.3 SLE has a clinical variant similar to erythema pernio (chilblain lupus), but differs in histology. It is worth clarifying the semantic confusion with lupus pernio, which is a cutaneous manifestation of sarcoidosis and is not related to lupus erythematosus or temperature changes.3,4 Until 2008, 70 cases of lupus perniosis had been reported, most with the sporadic presentation; however, there is a description of genetic association in two families.5
Case presentationA 57-year-old woman presented to the emergency room with a three-month history of simultaneous appearance of erythematous, nodular, and painful lesions on the hands, ears, and scalp, progressively increasing in size over the past month, with ulcers in some of the lesions. The patient also refers asthenia, adynamia, hair loss, xerophthalmia, polyarthralgia, asymmetric edema of the lower limbs, and episodes of fever of up to 39 °C. She was diagnosed with SLE, rheumatoid arthritis, nephrotic syndrome, and chronic kidney disease stage 2 secondary to class IV lupus nephritis. She was under treatment with chloroquine phosphate 250 mg/day and losartan 50 mg/day. Consulting with dermatology the previous month, they added 1% topical hydrocortisone, mycophenolate mofetil 250 mg/day, and prednisone 5 mg/day.
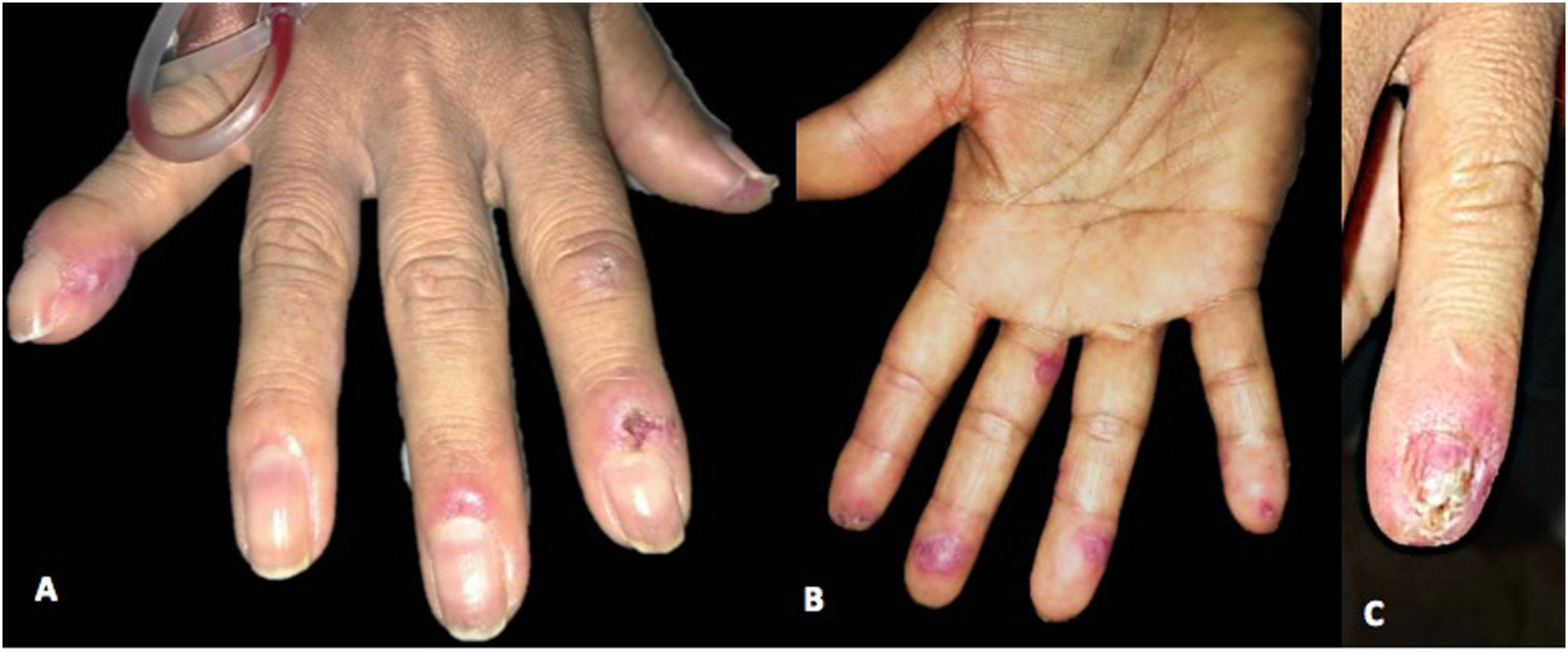
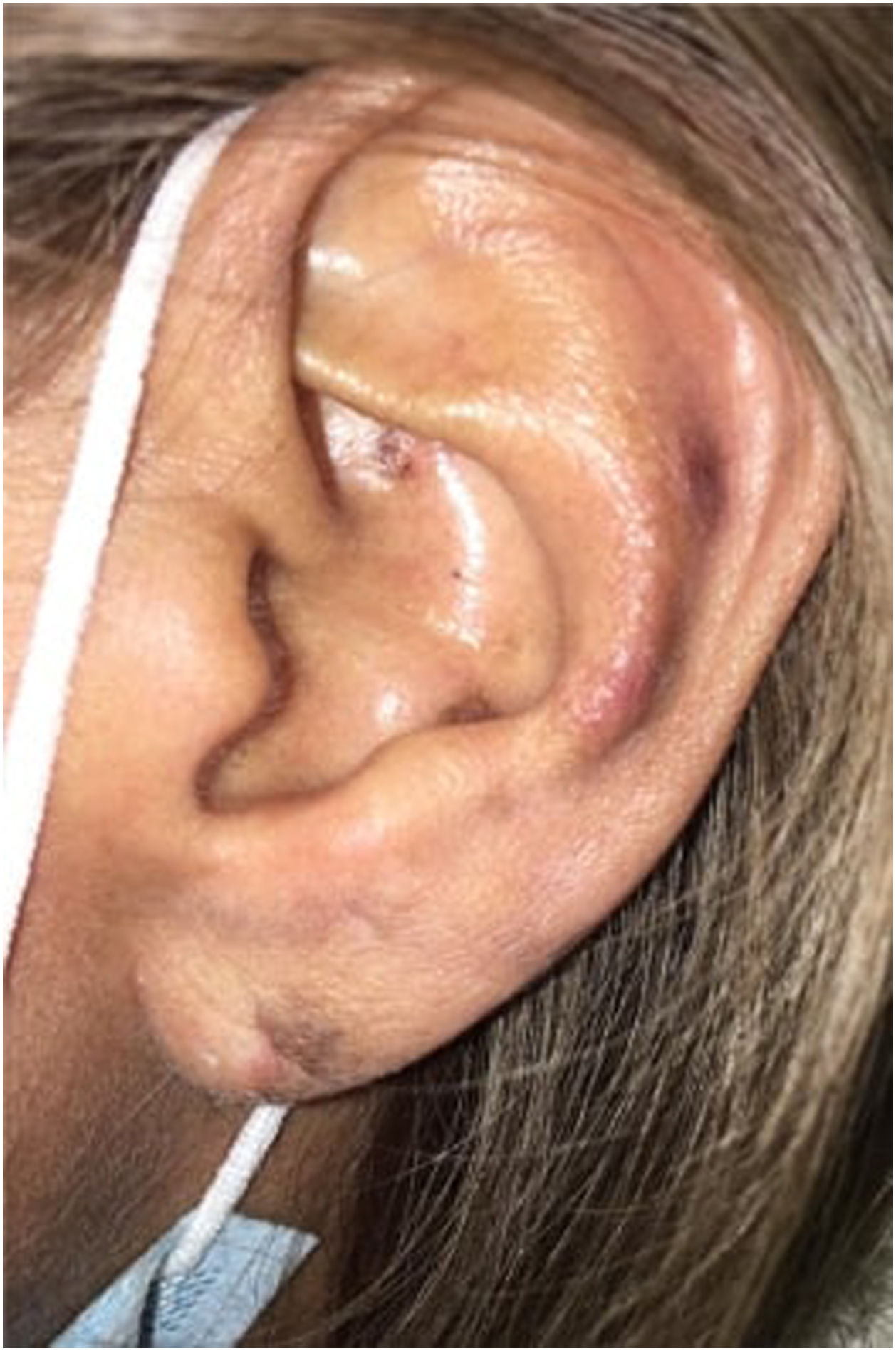
Physical examination depicts erythematous and violaceous plaques with well-defined irregular borders in the frontal, left parietal, occipital region of the scalp and left ear, pads, and proximal nailfold of both hands, and pads on the toes. In addition, she presented an ulcer with stellate configuration on the proximal nailfold of the second finger of the right hand, onycholysis changes and nail plate dystrophy in the fifth finger of the left hand, as well as multiple splinter hemorrhages in the nail plate of all the fingers of both hands and an indurated erythematous-violaceous nodule in the left ear (Figs. 1 and 2).
 In the proximal and lateral nail folds, fingertips, and palmar aspect of the left hand, erythematous-violaceous plaques with poorly defined irregular borders are observed. (C) In the fifth nail of the left hand, on the proximal nail fold, an erythematous-violaceous plaque, with nail dystrophy of 100% of the nail plate, distal onycholysis, and splinter hemorrhages are evidenced.")
(A and B) In the proximal and lateral nail folds, fingertips, and palmar aspect of the left hand, erythematous-violaceous plaques with poorly defined irregular borders are observed. (C) In the fifth nail of the left hand, on the proximal nail fold, an erythematous-violaceous plaque, with nail dystrophy of 100% of the nail plate, distal onycholysis, and splinter hemorrhages are evidenced.

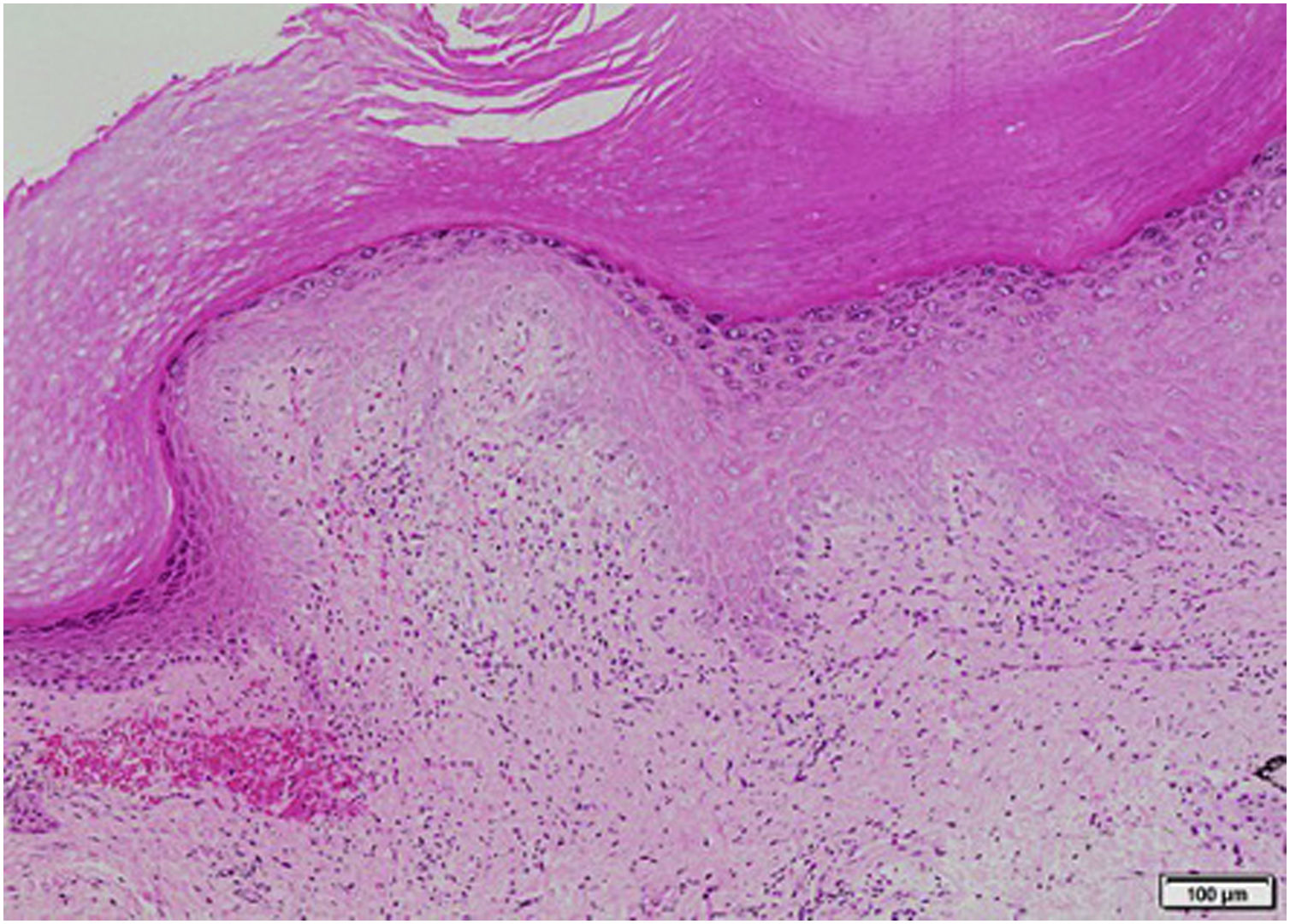
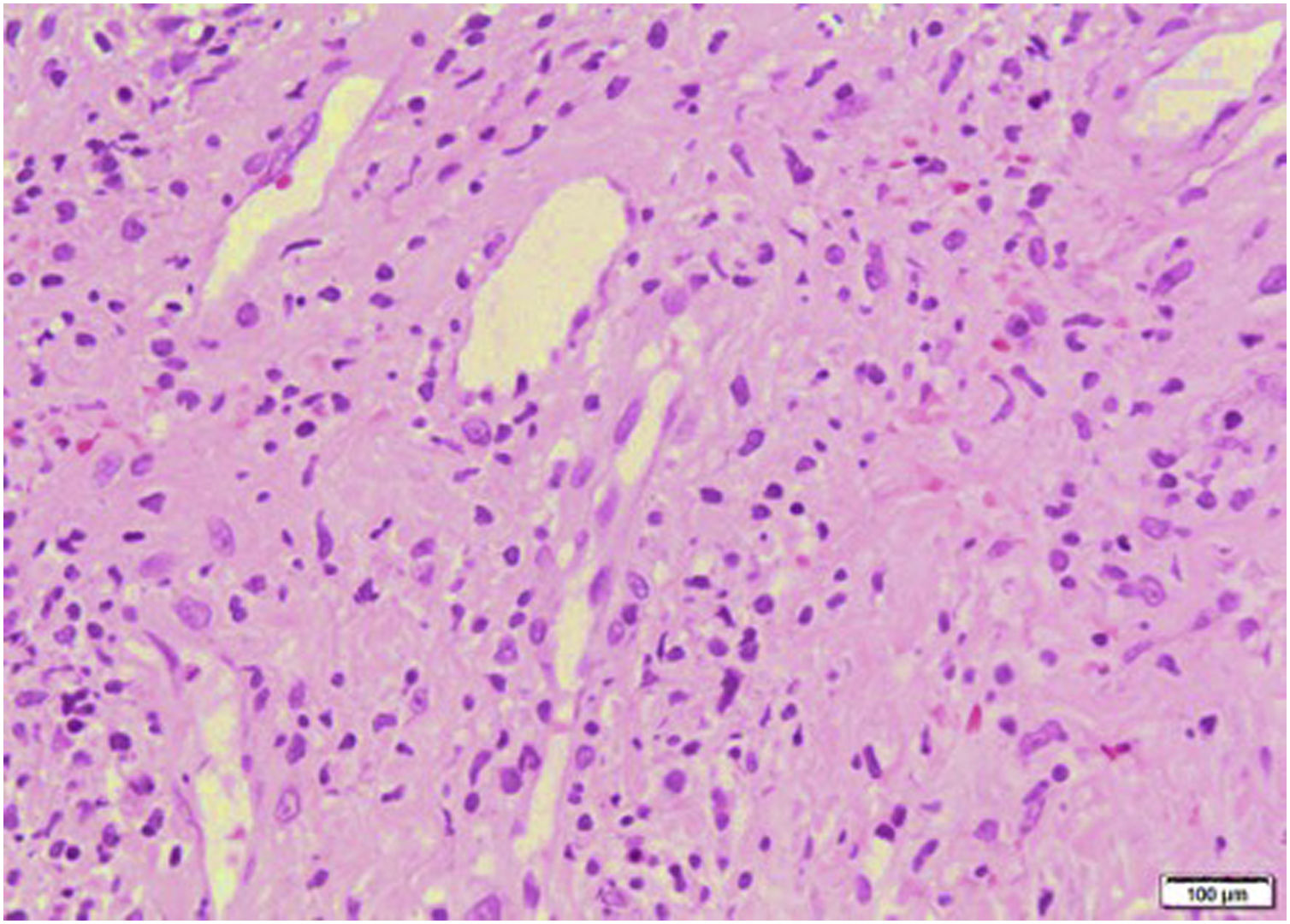
Lab test results can be seen in Table 1. Lower limbs Doppler analysis reported an acute deep vein thrombosis of the left popliteal vein. Antiphospholipid antibodies were negative. Hence, anticoagulation was started with enoxaparin followed by warfarin. Skin biopsies were taken from the ear and left hand, which reported findings suggestive of dermal-epidermal interface damage and perivascular lymphoid infiltrates (Figs. 3 and 4). Direct immunofluorescence demonstrated granular deposits of IgM, C3, and C1Q in the basement membrane. Clinical and histological findings were compatible with chilblain lupus, according to the Mayo Clinic criteria.6 Prednisone dose was increased to 1 mg/kg/day, with adequate response and remission of skin lesions.
Lab tests.
| Complete blood count | Leukocytes: 2,500/mm3, neutrophils: 1,400/mm3 (55%), lymphocytes: 800/mm3 (33%). Hemoglobin: 13.3 g/dL, MCV: 91.3 fL, MHC: 30.9 pg, Platelets: 129,200/mm3 |
| Renal function | Creatinine 0.79 mg/dL; blood urea nitrogen 16.7 mg/d. Urinalysis: pH 7.0, density 1.015, negative for proteins, leukocytes, and blood |
| Electrolytes | Sodium 137 mmol/L. Potassium 3.7 mmol/L. Chloride 101 mmol/L. Non-ionized calcium 8.4 mmol/L |
| Autoimmune profile | ANA: 1/320 homogeneous pattern; anti-ENA: negativeComplement: C3 43.4 mg/dL (88−165 mg/dL), C4 10.1 mg/dL (14−44 mg/dL)Lupus anticoagulant: negativeIg G anti-β2 glycoprotein antibodies: negativeIgM anti-β2 glycoprotein antibodies: negativeIgG anticardiolipin antibodies: negativeIgM anticardiolipin antibodies: negativeCold agglutinins: negative |
| Others | BNP: 59.8 pg/mL CK: 36 u/L, albumin 3.2 g/dL, aerobic and anaerobic blood cultures: negative |
MCV, Mean corpuscular volume; MHC, Mean hemoglobin concentration; ANA, Antinuclear antibodies; ENAS, Extractable nuclear antinuclear antibodies; Ig, Immunoglobulin; BNP, Brain natriuretic peptide; CK, Creatine phosphokinase.


SLE is associated with a wide variety of skin diseases.The spectrum of CLE is heterogeneous, thus Gilliam et al. proposed a classification based on the stratification of patients with similar clinical, histological features, and response to treatment.7 This classification comprises three morphological groups: acute, subacute, and chronic, the latter being the least related to systemic involvement.8 The group of CLE includes discoid, tumidus, hypertrophic, mucosal, profundus, and chilblain lupus.9–11 However, Gillian's classification has several limitations, such as chronic cutaneous lupus lesions could not be classified histologically into any of the morphological types; thus, Lipsker proposed a new classification that divided the clinical manifestations of lupus into specific and non-specific.11
The histological characteristics of the specific lupus lesions include the presence of interface dermatitis, vacuolization, necrosis of basal keratinocytes, thickening of the basement membrane, incontinence of pigment, and lymphocytic infiltrate in the dermo-epidermal junction.7 The distinct signs of CLE are subdivided according to the level of histological involvement. Acute, subacute, chronic, and vesiculobullous cutaneous lupus are expressed by dermal-epidermal affection. Lupus tumidus, Jessner's lymphocytic infiltrate, and erythematous and papulo-nodular reticular mucinosis secondary to SLE are characterized by dermal affection, while lupus panniculitis alters subcutaneous cellular tissue.11,12
Non-specific signs of lupus are also classified according to their pathogenesis; for instance, thrombotic vasculopathy comprises Degos papules, livedo racemosa, atrophie blanche, purpura, splinter hemorrhages, skin necrosis, anetoderma, and thrombophlebitis; cutaneous neutrophilic manifestations of lupus erythematosus includes bullous lupus, urticarial vasculitis, and neutrophilic dermatoses (including amicrobial pustulosis of the folds, neutrophilic urticarial dermatosis, Sweet's syndrome, and pyoderma gangrenosum). Finally, there are diseases with uncertain pathogenic significance, such as rheumatoid nodules, interstitial granulomatous dermatitis, and eruptive fibromas.11,12
Chilblain lupus is an uncommon form of chronic CLE defined by lesions in acral areas exposed to cold, causing vasoconstriction and microvascular injury10; most cases are sporadic and often affect middle-aged women; an association with low body mass index and positive anti-Ro antibodies has been described.13–15 The pathophysiology is not fully understood, but it is autoimmune and is related to altered microcirculation, stasis, and vascular occlusion precipitated by low temperatures. There are hypotheses concerning the migration of the Ro antigen from the nucleus to the membrane of the keratinocyte due to the physical stimulus of cold.14,15
Familial cases have been described in which chilblain lupus is known as a genodermatosis with an autosomal dominant inheritance pattern. Causal genes that encode intracellular nucleases such as TREX1, SAMHD1, and STING3,16 are included, which produce the activation of type 1 interferon, which, in turn, leads to the immune recognition of non-metabolized nucleic acids5,16,17. In the hereditary pattern, the lesions are also induced by cold, associated with arthralgia, positive antinuclear antibodies (ANA), or lymphopenia.3
The typical clinical lesions are painful erythematous-violaceous macules, plaques, and nodules, usually in exposed areas, such as the back of the hand and the pads of the fingers or toes, and rarely on the tip of the nose or the ears. There may be central erosions or ulcers.9,10 Infrequently, hyperkeratosis with fissures can be observed on the soles and palms.14 Cases of nail involvement with dystrophy or loss of the nail plate have been described, indicating chronic changes, as observed in the clinical case described here.14 Moreover, patients with chilblain lupus may present with hypergammaglobulinemia (2/3 of cases), and high-titers of rheumatoid factor, ANA, and anti-Ro antibodies.5,10 There is a relationship with an increase in red blood cells, and this could cause blood viscosity and stasis; magnetic resonance angiography studies have shown decreased perfusion in the phalanges in these cases.
The diagnostic criteria for this entity proposed by Su et al.6 (Table 2) include the presence of two major criteria and at least one minor criterion. The histopathology of chilblain lupus consists of epidermal atrophy, interface vacuolization, papillary edema, and perivascular mononuclear infiltrate in the dermis. Direct immunofluorescence shows deposition of IgM, IgA, or C3 at the dermal-epidermal junction, along with perivascular deposits of C3 and fibrinogen.5,16 The presence of eccrine periadnexal distribution of lymphocytic infiltrate is observed in the idiopathic form (not related to lupus) and helps in their differentiation.3 The current case has the classic histological findings and meets diagnostic criteria.6
Mayo Clinic diagnostic criteria for chilblain lupus.6
| Reported patient | ||
|---|---|---|
| Major criteria | Acral skin lesions induced by exposure to cold or temperature drops | Acral lesions (hands, feet, and ears) induced by exposure to cold |
| Evidence of skin lesions secondary to lupus erythematosus, determined by histopathological assessment (hyperkeratosis, vacuolated basement membrane, and predominantly lymphocytic perivascular or periadnexal infiltrate; in chronic cases, atrophic epidermis and thickening of the basement membrane) or direct immunofluorescence (granular deposition of IgM, C3, or IgG in the basement membrane) | The skin biopsy was compatible with epidermal atrophy with vacuolar damage of the dermal-epidermal interface and dyskeratocytes. In addition, the presence of edema and perivascular lymphoid infiltrates with lymphocytes permeating the blood vessel wall, without evidence of necrosis | |
| Minor criteria | Coexistence of systemic lupus erythematosus or other discoid lupus erythematosus skin lesions | Positive ANA (1:320 with speckled pattern), consumed complement, and history of lupus nephritis confirmed by biopsy. |
| Response to treatment for lupus erythematosus | An adequate response to prednisone at a dose of 1 mg/kg/day | |
| Negative tests for cryoglobulins and cold agglutinins | A negative result for cryoglobulins |
Treatment of chilblain lupus is not standardized. The first line described for localized CLE is high-potency topical steroids, such as 0.05% clobetasol, combined with physical measures to avoid cold.9,18 In some cases, the adequate response has been obtained in subjects treated with calcium channel blockers, such as nifedipine at 60 mg/day, to counteract the phenomenon of vasoconstriction.5,18 The use of chloroquine or hydroxychloroquine can be considered, but the response is only achieved in approximately one in three individuals.18,19 Another therapeutic alternative is topical pimecrolimus or tacrolimus. Cases of induction of improvement in lesions have been reported with mycophenolate mofetil at a dose of 2–3 g/day in patients with clinical worsening, despite hydroxychloroquine, prednisone, and other topical agents.20,21
In case of extensive or refractory to topical treatment lesions, the use of systemic steroids, chloroquine, or hydroxychloroquine5,9 is suggested. In the reported case, the dose of mycophenolate mofetil was lower than that recommended20,21; this fact is associated with progression of the lesions, so the treatment of choice was prednisolone at 1 mg/kg per day to achieve clinical remission.
Biologics have been used for the management of lupus erythematosus. In patients with SLE, the proportion of precursor, transitional, memory B cells, and plasmablasts is increased, as well as the expression of co-stimulatory molecules such as CD86 in CD19+ cells.22–25 These cells play a central role in the pathogenesis due to the loss of cellular tolerance and the production of antibodies against nuclear DNA and nucleosome complexes.26 Rituximab is a chimeric monoclonal antibody against the CD20 molecule on the surface of B lymphocytes and induces a response in subjects with lupus nephritis and non-renal manifestations refractory to systemic treatment.27–29 Vital et al. carried out a prospective study with 26 patients, in which 42.6% of the individuals with acute cutaneous lupus responded29; however, there was no response in patients with CLE, and, paradoxically, nine patients without skin manifestations developed subacute cutaneous lupus during follow-up after administration of rituximab.29 This could suggest the different roles of the B cell in the pathogenesis of the different types of lupus.25,27–29
Another biologic used to treat the cutaneous manifestations of lupus is belimumab, a monoclonal antibody that inhibits the immunomodulatory cytokine (BLyS or BAFF), which stimulates the differentiation and survival of B lymphocytes.25 A pooled analysis of two phase-III trials of belimumab (BLISS-52 and BLISS-76) at doses of 1 and 10 mg/kg applied on days 0, 14, and 28, and every four weeks thereafter, compared to standard therapy, demonstrated improvement of mucocutaneous manifestations (rash, mucosal ulcers, and alopecia) in patients with SLE.27 Improvement was defined as recovery of more than 50% of the lesions and a diminution in the steroid dose by 25%; this outcome was achieved in 43% and 52% of the patients with mucocutaneous manifestations, at three and six months, respectively.28 Another recent study of 50 subjects with SLE with an indication for treatment with belimumab at a dose of 10 mg/kg found a 12-month probability of achieving low disease activity status and remission of 58.1% and 37.1%, respectively30; in the latter group, 10% of the population had active cutaneous lupus with the erythema pernio variant.30 Treatment with tofacitinib, a small-molecule synthetic JAK inhibitor that suppresses interferon 1 signaling, has been described for familial chilblain lupus with heterogeneous STING gene mutation, with a reduction of pain and lesions with a dosing schedule of 5 mg every 12 hours for 17 days.17
ConclusionThis is a case report where the diagnosis and treatment of SLE preceded the cutaneous manifestation of chilblain lupus with nail involvement. The patient responded adequately to systemic steroids and remission of the skin lesions was achieved at follow-up. Although perniosis in lupus erythematosus does not respond well to antimalarials, physical measures of avoiding cold, use of calcium-channel blockers, and topical or systemic steroids were associated with good results. In refractory cases, other immunomodulators can be considered, such as mycophenolate mofetil or biologics.
Ethical considerationsThe authors declare that this work received the approval of the Research and Institutional Ethics Committee of the Faculty of Medicine of the Pontificia Universidad Javeriana. Likewise, the authors confirm that the informed consent of the patients was obtained and that at all times they have preserved the confidentiality of their data.
Conflict of interestsThe authors declare that they have an absence of conflict of interest.
Please cite this article as: Tamayo Buendía MM, Ordóñez-Parra J, Moreo-Mercado S, Mejía Cortés M. Perniosis lúpica con compromiso ungueal: un reporte de caso. Rev Colomb Reumatol. 2022;29:231–236.
