



Granulomatosis with polyangiitis is a systemic vasculitis that affects medium and small vessels, with high expression of anti-neutrophil cytoplasmic autoantibody. A case is presented on a patient with an initial compromise of the lower airway, who did not respond to management, required intensive care unit management, and died due to severe diffuse alveolar hemorrhage. His definitive diagnosis was established with a clinical autopsy. Granulomatosis with polyangiitis is a disease with different ways of presentation, and can have fatal outcomes if it is not diagnosed early.
La granulomatosis con poliangeítis es un tipo de vasculitis que afecta a vasos de mediano y pequeño calibre de manera sistémica, con una alta expresión de anticuerpos contra el citoplasma del neutrófilo. Se presenta el caso de un paciente con un compromiso inicial de la vía área inferior, que no respondió al tratamiento y requirió manejo en unidad de cuidados intensivos. Finalmente, falleció por una hemorragia alveolar difusa severa. Su diagnóstico definitivo se estableció con una autopsia clínica. La granulomatosis con poliangeítis tiene diferentes formas de presentación y puede tener desenlaces fatales si no se diagnostica a tiempo.
Granulomatosis with polyangiitis (GPA) is a complex systemic vasculitis-like disease that affects vessels of medium and small caliber.1 It is known by the eponym of Wegener's disease, in honor of its first describer, the German pathologist Friedrich Wegener.2 This pathology has a complex multifactorial etiology, low incidence and prevalence, and occurs in different forms. The diagnosis is usually made through a combination of clinical manifestations and laboratory tests, while treatment depends on several factors that influence the outcome, as well as the progression of the disease.3
This article is a case report of a 16-year-old male patient diagnosed with GPA, with an early and severe systemic presentation, who was admitted with symptoms suggestive of pneumonia associated with hemoptoic expectoration, and did not respond to different lines of antimicrobial management during his hospitalization, for which he required management in the intensive care unit (ICU) due to ventilatory failure. The final diagnosis of GPA was made with a clinical autopsy 16 days after the onset of symptoms.
The objective of this article is to record and analyze the clinical case of a patient with GPA, which had a systemic and early presentation that was fulminant.
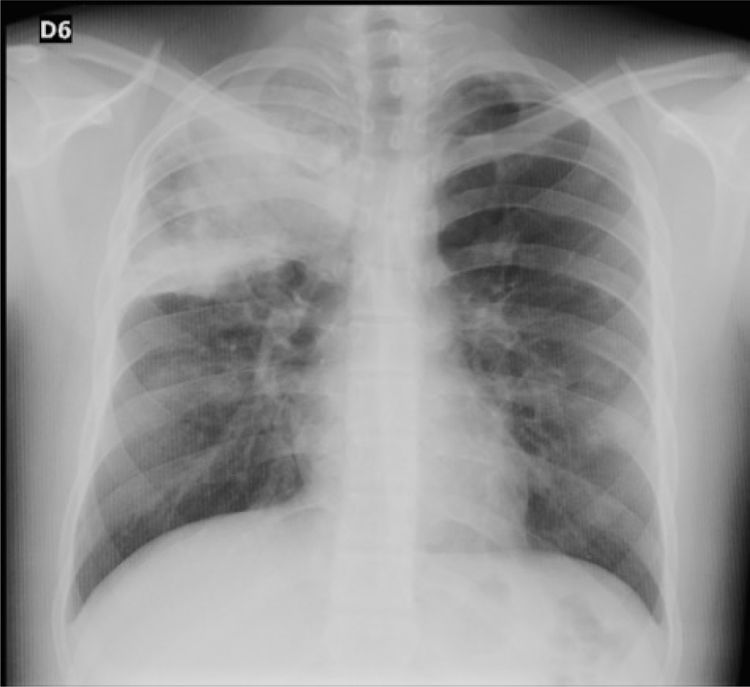
Case reportA 16-year-old male patient with a history of allergic rhinitis and conjunctivitis, hyperhidrosis managed with bilateral sympathectomy and a maternal history of hypothyroidism, who consulted due to symptoms of 10 days of evolution of hyaline rhinorrhea, nasal congestion, odynophagia, non-productive cough, general malaise, global headache, epistaxis and fever quantified in 39 ° C. On admission, he presented the following vital sign values: heart rate of 121, weight of 67 kg, height of 161 cm, BMI of 25.8 kg/m2, without abnormal lung sounds, oxygen saturation of 95%, and no other findings on physical examination. The chest X-ray showed a consolidation in the right upper lobe (Fig. 1), for which management for community-acquired pneumonia was started, on an outpatient basis, with oral erythromycin for 7 days.

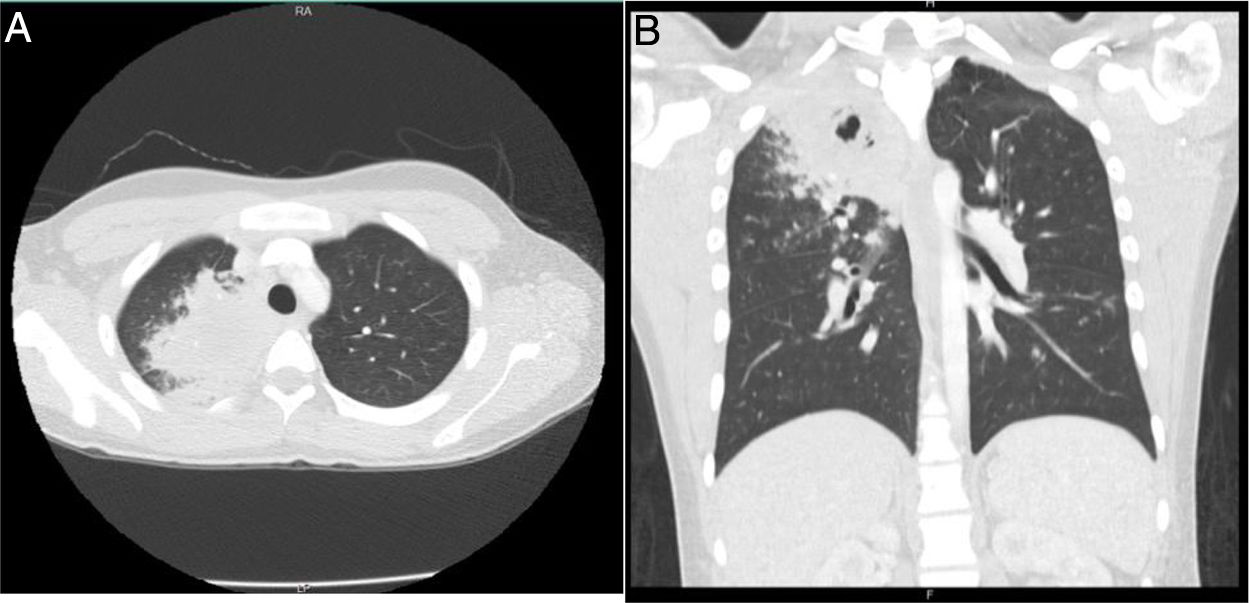
24 h after, the patient was readmitted due to persistence of the symptoms, to which nausea, vomiting, abdominal pain, and dyspnea were added, and for this reason was managed with nebulized bronchodilators, intravenous fluids, metoclopramide, and intravenous dipyrone, with partial improvement in symptoms. Later, he presented clinical deterioration, with the appearance of hemoptysis, foul-smelling stools, vital signs with blood pressure of 112/57 mmHg, heart rate of 109, respiratory rate of 18, temperature of 36.8 ° C, oxygen saturation of 96%, appearance of fine rales in the right apex, without lesions in the nostrils, significant leukocytosis (16,680), eosinophilia (11.4%) and 9% bands (Table 1). For this reason, he was hospitalized and empirical management with ampicillin sulbactam and clarithromycin was initiated. The Gram stain and the sputum culture were negative, the chest tomography showed parenchymal opacities that compromised the apical segment of the right upper lobe and micronodular images that formed a "tree-in-bud" with minimal central necrosis (Fig. 2). New blood and stool cultures were taken, which were negative. The patient presented a new prolonged febrile peak that was difficult to manage, so vancomycin was started. Bronchoalveolar lavage by bronchoscopy was performed, in which potassium hydroxide, Koch's bacillus and bacteria were negative.
Laboratory data.
| Variables | Reference values | Admission | Day 5 | Day 6 | Day 8 | Day 11 | Day 14 |
|---|---|---|---|---|---|---|---|
| Hemogram | |||||||
| Leukocytes (cells/uL) | 5,000-10,000 | 16,688 | 14,880 | 13,570 | 10,410 | 17,340 | 27,800 |
| Neutrophils (cells/uL) | 36.7-64,1 | 12,090 (72.5%) | 11,530 (77.5%) | 11,210 (82.6%) | 8,670 (83.3%) | 13,720 (79.1%) | 23,510 (84.6%) |
| Lymphocytes (cells/uL) | 21.2-39.7 | 1600 (9.6%) | 880 (5.9%) | 750 (5.5%) | 1150 (11%) | 2180 (12.6%) | 3070 (11%) |
| Monocytes (cells/uL) | 4.05-12.8 | 1050 (6.3%) | 1050 (7.1%) | 610 (4.5%) | 560 (5.4%) | 870 (5.0%) | 970 (3.5%) |
| Eosinophils (cells/uL) | 1-3.9 | 1900 (11.4%) | 1370 (9.2%) | 960 (7.1%) | 0.0 (0%) | 540 (3.1%) | 220 (0.8%) |
| Basophils (cells/uL) | 0.01-1 | 40 (0.3%) | 50 (0.4%) | 40 (0.3%) | 30 (0.2%) | 30 (0.2%) | 30 (0.2%) |
| Bands (%) | 0 | 9 | 3 | 0 | 0 | 0 | 0 |
| Hemoglobin (g/dl) | 14-18 | 11.9 | 10.5 | 12.1 | 11.3 | 10.1 | 7.8 |
| Hematocrit (%) | 45-56 | 38.5 | 33.9 | 38.1 | 34.2 | 30.7 | 23.5 |
| Mean corpuscular volume (fl) | 80-100 | 83.9 | 82.9 | 82.1 | 80.7 | 81.6 | 82.5 |
| Mean corpuscular hemoglobin (pg) | 27-34 | 25.9 | 25.7 | 26.1 | 26.7 | 26.9 | 27.4 |
| Platelets (cells/uL) | 150,000-450,000 | 579,000 | 506,000 | 589,000 | 911,000 | 744,000 | 575,000 |
| Blood chemistry | |||||||
| C reactive protein (mg/dl) | < 6 | 12 | 48 | 24 | |||
| Procalcitonin (ng/ml) | 0-0.500 | 0.218 | 0.52 | 0.56 | |||
| Ureic nitrogen (mg/dl) | 5.1-18 | 17.1 | 10 | 23.8 | 21.7 | ||
| Creatinine (mg/dl) | 0.67-1.17 | 0.78 | 0.85 | 0.77 | 1.17 | ||
| Transaminase ALT/TGP (u/l) | < 26 | 135.6 | 70.2 | ||||
| Transaminase AST/TGO (u/l) | 26-31 | 49.1 | 24.1 | ||||
| Albumin (g/dl) | 3.50-5.0 | 3.2 | 2.6 | ||||
| Fibrinogen (mg/dl) | 0-339 | 462 | |||||
| Uric acid (mg/dl) | 2.1-7.6 | 4.6 | |||||
| Urine cytochemistry | |||||||
| Proteins (mg/dL) | Negative | Negative | Negative | 25 | 75 | ||
| Leukocyte/ esterase | Negative | Negative | Negative | Negative | Negative | ||
| Erithrocytes (cells /uL) | Negative | 25 | 250 | 250 | 250 | ||
| Urine sediment | |||||||
| Red blood cells (cells x field) | Negative | 4 xC | >20 xC | >20 xC | >20 xC | ||
| Fresh | Negative | 50% | 20% | 20% | |||
| Crenated | Negative | 50% | 30% | 80% | |||
| Immunologic | |||||||
| Immunoglobulin E (IU/ml) | 0-60 | 312 | 99.5 | ||||
| Immunoglobulin A (IU/ml) | 90-310 | 105 | |||||
| Immunoglobulin G (IU/ml) | 710-1520 | 975 | |||||
| Immunoglobulin M (IU/ml) | 40-157 | 73 | |||||
| Complement C3 (mg/dl) | 90-180 | 175.1 | |||||
| Complement C4 (mg/dl) | 10.0-40 | 28.1 | |||||
| ANCA-MPO (U/ml) | 11.0-18 | 0.6 | |||||
| ANCA-PR3 (U/ml) | 11.0-18 | 238.2 | |||||
ALT/TGP: alanine aminotransferase/glutamic pyruvic transaminase; ANCA: anti-neutrophil cytoplasmic antibody; ANCA-MPO: ANCA against myeloperoxidase; ANCA-PR3: anti-proteinase 3 ANCA; AST/TGO: aspartate aminotransferase/glutamic oxaloacetic transaminase.
 axial section. B) Coronal section. Parenchymal opacity in the apical segment of the right upper lobe, with areas suggestive of cavitation with micronodular images, which configure a \"tree-in-bud\".")
After 5 days, the patient presented an inadequate response to management, a complement test was requested that showed increased C3, with positive ANCA-PR3 (anti-neutrophil cytoplasmic proteinase 3 antibody), and persistence of leukocytosis (14,880) and eosinophilia (9.2%) was detected in the blood count. A tuberculin skin test (PPD) which was negative, a CT scan of the paranasal sinuses which was normal and new blood cultures which were negative were performed. Eosinophilic granulomatous vasculitis vs. granulomatosis with polyangiitis (Wegener's disease) were suspected; intravenous corticosteroids were started and the antibiotic treatment was changed to ceftriaxone with clindamycin. The patient presented a new febrile peak, the antibiotic was escalated again to vancomycin, and ceftriaxone was continued. The control blood cultures were negative, leukocytosis persisted (17,000) and thrombocytosis started (744,000). The patient presented epigastric abdominal pain and emetic episodes, for which omeprazole was started, with no improvement, and it was taken an abdominal ultrasound that showed hepatomegaly and diffuse alteration of renal echogenicity, but with preserved renal function.
Sixteen days after admission, the patient presented poor general condition, grade 2 dehydration, generalized pallor, heart rate of 135, and oxygen saturation of 92% with FiO2 at 50%. Likewise, he was polypneic, with supraclavicular retractions, decreased vesicular murmur, decreased hemoglobin (7.8) and frank leukocytosis (27,800). A new chest X-ray showed generalized alveolar infiltrates with a predominance of peripheral formation, which is why it was considered that the patient had a diffuse alveolar hemorrhage vs. adult respiratory distress syndrome, for which he was transferred to the ICU. Already in the unit, the patient presented deterioration in his respiratory pattern, oxygen saturation of 82% with FiO2 at 50%, severe coughing fits with frank hemoptysis, and persistent respiratory distress, for which orotracheal intubation was performed and mechanic ventilation was started. The urinalysis showed proteinuria and hematuria, and the coagulation times were normal; he also had hypoproteinemia and hypoalbuminemia. Packed red blood cells were transfused, a viral panel and a new sputum smear and blood culture were performed, which were negative. The transthoracic echocardiogram showed moderate to severe pulmonary hypertension (psap = 72 mmHg), with hypertrophy and dysfunction of the right ventricle, for which treatment was started with dobutamine, norepinephrine and intravenous cyclophosphamide with corticosteroid pulses. A new fibrobronchoscopy was performed and moderate hemoptysis was evidenced. The patient continued to deteriorate, so management with milrinone was added, without improvement, and plasmapheresis was started. In addition, he presented pulmonary-renal syndrome, with non-oliguric renal failure and severe diffuse alveolar hemorrhage. Plasma and albumin replacement was started, but the patient did not respond to management and the oxygenation disorder persisted.
Three days after admission to the ICU, the patient presented progressive bradycardia with hypotension, without response to vasopressors at maximum doses. Marked desaturation (40%) persisted with maximum ventilatory support; successively he presented 3 episodes of cardiac arrest, which responded to management with adrenaline and chest compressions. In the last event he presented asystole that did not respond to management, advanced cardiopulmonary resuscitation was performed, but the presence of vital signs was not achieved, so death was declared.
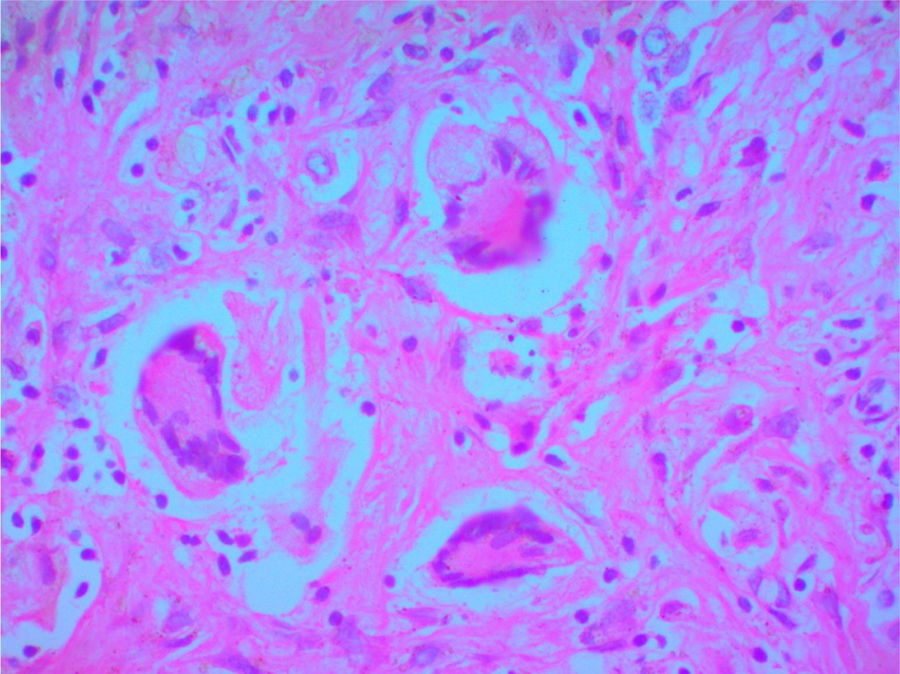
A clinical autopsy was requested due to an unclear cause of death, and at the macroscopic level, an irregular lesion with induration and an area of necrosis on the surface was found in the upper right lobe of the lung. Multiple nodular lesions were observed in the lung parenchyma and a hemorrhagic appearance in the rest of it. At the microscopic level, the lung parenchyma presented alveolar hemorrhages and areas of necrosis (Fig. 3), in addition to granulomas surrounded by an area of fibroblastic proliferation with leukocyte infiltrate and giant multinucleated cells with obliterated vessels due to vasculitis. Special Periodic Acid-Schiff, Ziehl-Neelsen and Gromory stains were performed, which were negative for microorganisms and positive for fibrinogen in the alveolar spaces. These findings confirmed the diagnosis of GPA (Wegener's granulomatosis).
Discussion
GPA is an ANCA-associated systemic disease, defined by the international Chapel Hill consensus as a necrotizing vasculitis with involvement of vessels of small to medium caliber, associated with granulomatous inflammation that can compromise the upper and lower airways and the kidney. with glomerulonephritis and focal necrosis.4 This pathology has an estimated incidence in North America of 10.8 new cases per million people/year (95% CI: 10.7-12.9),5 and in South America of 9 cases per million people/year, with a prevalence of 7.4 per 100,000 inhabitants (95% CI: 2.8-12).6 Its presentation in the pediatric population is higher in women (70%)7; however, in our case it presented in a young man, 16 years old, with no risk factors or important antecedents.
From the beginning, the clinical manifestations presented corresponded to a compromise of the lower airway, with a slight involvement of the upper airway (hyaline rhinorrhea),8 which demonstrates the heterogeneity in its presentation, since the commitment reported in clinical studies usually consists of kidney involvement, constitutional symptoms (fever, weight loss, myalgia, among others) and otorhinolaryngological manifestations.9 Initially, the poor response to the use of antimicrobials should have indicated that it was not an infectious pathology. Multiple blood cultures, direct cultures of bronchoalveolar lavages and sputum samples did not isolate any germ. However, it has been determined that infections with bacteria such as Staphylococcus aureus may suggest a possible etiology of this vasculitis.10 There are other studies that describe different risk factors, such as decreased exposure to ultraviolet rays or greater latitude, which could predispose to presenting this disease,11 as well as the antecedent of use of certain medications such as antithyroid drugs or antibiotics, can favor the production of ANCA by polyclonal B lymphocytes.12
As for the management received, the start of intravenous corticosteroids could condition the response to treatment, obtaining a plateau in the progression of the disease, since it is within the lines of management proposed by the European League Against Rheumatism (EULAR), but no response was seen when boluses of cyclophosphamide were used, nor was any change seen when plasmapheresis was performed.13 The use of rituximab, a monoclonal antibody against the CD-20 antigen of B lymphocytes,14 was considered, but the inadequate response and the presence of diffuse alveolar hemorrhage shortened his survival, so management with this drug could not be attempted.
Given the heterogeneous clinical manifestations, the poor clinical evolution and the poor response to pharmacological management, with a fatal outcome for the patient, it was decided to confirm this diagnosis with a clinical autopsy, since the presence of fever during hospitalization prevented from performing a lung biopsy to confirm the GPA. Finally, the pathology report showed the existence of granulomas with areas of necrosis in alveolar spaces, which demonstrated that it was indeed a GPA, but with a fulminant early systemic presentation.
ConclusionsGPA is a complex multifactorial pathology, with various clinical manifestations, and specific criteria have not been unified to be able to issue an early diagnosis and timely start of treatment. The study Diagnostic and Classification Criteria in Vasculitis, an observational study that will unify these criteria and will allow adequate early management of patients is being conducted at this time.15 The early and severe forms of presentation influence the prognosis of the disease, and therefore its mortality, so clinical trials that inquire into this type of presentation are required.16 The treatment suggested by the EULAR is in force, and the follow-up must be carried out in accordance with the proposed guidelines.14 The objective in the future for current medicine will be to make a timely and early identification of this type of cases, in order to initiate early management of the disease and achieve a significant impact on survival and thus improve its prognosis.
Ethical considerationsThe case has informed consent in which the use of information and disclosure of the clinical data of the patient is authorized.
Conflict of interestThe authors declare that they have no conflict of interest.
The authors would like to thank the Department of Pathology of the Universidad de la Sabana for their support, especially Dr. Néstor Segundo Beleño Beltrán and Dr. María Claudia Abaúnza Chaguin, for digitizing the images obtained.
The following is Supplementary data to this article:
Please cite this article as: Hernández-Acosta R, Bastidas-Goyes A, Hernández-Rincón E. Granulomatosis con poliangeítis, reporte de caso de una presentación sistémica temprana fulminante. Rev Colomb Reumatol. 2022;29:225–230.
