




Hematological alterations are common in patients with systemic lupus erythematosus (SLE). These hematological manifestations may be expressed related to the involvement of cells affected and coagulation changes. The compromise in coagulation is associated with thrombotic manifestations. Risk factors associated with thrombosis have been described, such as the presence of elevated levels of homocysteine, acquired deficit of protein S, protein C, and antithrombin. However, the hemorrhagic diathesis has also been described at a lower frequency and related to the acquired deficiency of coagulation factors caused by the development of autoantibodies directed against coagulation factors. The cases are presented of 3 patients with juvenile SLE with unusual hematological manifestations, as well as a review of the literature in relation to them.
The hematological manifestations in juvenile SLE are not only related to alterations in cell lines, vascular thrombosis and bleeding disorders should also be suspected. Early diagnosis and treatment reduces morbidity and mortality related to this type of manifestations.
Las alteraciones hematológicas son comunes en los pacientes con lupus eritematoso sistémico (LES). Pueden expresarse relacionadas con el compromiso de las líneas celulares y con la presencia de alteraciones de la coagulación. El compromiso en la coagulación se asocia con manifestaciones trombóticas. Se han descrito factores de riesgo asociados a trombosis, como la presencia de niveles elevados de homocisteína, déficit adquirido de la proteína S, proteína C y antitrombina. Sin embargo, la diátesis hemorrágica también se ha descrito con menor frecuencia y relacionada con el déficit de factores de la coagulación, secundaria a la presencia de inhibidores. Presentamos 3 pacientes con LES juvenil con manifestaciones hematológicas poco usuales y revisión de la literatura relacionada.
Se concluye que las manifestaciones hematológicas en LES juvenil no solo se relacionan con alteraciones en las líneas celulares. Trombosis vasculares y trastornos hemorrágicos deben sospecharse. El diagnóstico precoz y el tratamiento temprano disminuyen la morbimortalidad relacionada con este tipo de manifestaciones.
Systemic lupus erythematosus (SLE) is considered the prototype of autoimmune disease associated with a component of autoinflammation. It is characterized by presenting multisystemic compromise and heterogeneous clinical manifestations in the course of the disease.1,2
The American College of Rheumatology (ACR) developed the first classification criteria in 1971, which were modified in 1982. Later, in 2012 the Systemic Lupus International Collaborating Clinics (SLICC) established a new group of criteria, which include 11 clinical and 6 immunological criteria. The presence of 4 criteria, among which there must be one clinical and one immunological or a renal biopsy with findings compatible with lupus nephritis associated with the presence of antinuclear antibodies (ANA) or anti-dsDNA antibodies is required.3 The ACR criteria have a sensitivity of 76.6% and a specificity of 93.4% unlike the SLICC criteria, which are more sensitive than specific: 98.7% and 85.3%, respectively.3 Hartman et al. published a meta-analysis, in which, based on the sensitivity and specificity for the pediatric population and in order to avoid false positives, they recommend the pediatric rheumatologists to use the ACR criteria.4
The incidence and prevalence of the disease are variable. It has been estimated a prevalence of 3.3–8.8 per 100,000 individuals of pediatric age and an incidence of 0.3–0.9 per every 100,000 individuals/year. The age of onset is variable, although it is more frequent between the ages of 12 and 16 years, with predominance in the female gender (4.5–5:1).1,5,6 The juvenile-onset SLE (jSLE) accounts for 10–25% of all cases of SLE.1–3
As for the clinical manifestations of jSLE, hematological commitment is a common finding, in different series its frequency ranges between 50 and 100% of patients and it can compromise the lineages of red cells, white cells, platelets or the coagulation system.2,5
The most frequent hematological alterations are in cell lineages. First, anemia is present in 80–90%, its etiology in descending order is anemia due to chronic disease, anemia due to iron deficiency and hemolytic anemia.1–3 In the latter, described in approximately 10–15%, it is noteworthy that 30–40% of patients have a positive direct Coombs test without other findings suggestive of hemolysis.3
Among the alterations of the white cell lineage, the most frequent is leukocytopenia: it occurs between 46 and 64% of patients with jSLE. It is possible to find neutropenia and lymphopenia. The latter is more prevalent and is considered a marker of disease activity.1–3 Finally, thrombocytopenia is present in 7–30% of patients and approximately in 15% of cases at the onset of the disease in pediatric age.3
The second clinical scenario in terms of hematological manifestations in jSLE consists of alterations in coagulation, which can be thrombotic or hemorrhagic; the thrombotic are more frequent. Although their frequency is not as high as that of alterations in cell lineages, they are associated with high morbidity and mortality.1–5 In this article we present 3 patients with jSLE with unusual hematological manifestations as well as a review of related literature.
Case reportsCase 1A 15-year-old male patient, diagnosed at the age of 13 years with antiphospholipid antibody syndrome (APS), which met criteria for deep vein thrombosis at the level of the right popliteal vein, with positive lupus anticoagulant and positive IgG and IgM anti-cardiolipin antibodies. The patient received anticoagulant management with low molecular weight heparins and chloroquine for 8 months, with treatment abandonment.
The patient was admitted to our service due to persistent fever and vasculitic lesions in lower limbs of a month of evolution. On physical examination, stage ii arterial hypertension, cervical and axillary adenomegalies, severe Raynaud's phenomenon with initial lesions of ulceration in the finger pads of hands and feet, and edema in lower limbs were found. The initial paraclinical tests showed leukocytopenia, lymphopenia, normochromic normocytic anemia, negative direct Coombs test, normal reticulocytes, ANA titer 1/160, anti-DNA titer 1/320, positive anti-RO and anti-La, positive lupus anticoagulant and IgG and IgM anticardiolipin antibodies, C3 and C4 hypocomplementemia, nephrotic-range proteinuria and venous Doppler ultrasound of the lower limbs with chronic deep venous thrombosis partially recanalized, which involved the right popliteal vein without findings suggestive of acute thrombi or arterial commitment. A diagnosis of SLE and APS was considered. Immunosuppressive management was started with pulses of methylprednisolone and cyclophosphamide associated with protective measures against cold, acetylsalicylic acid, antimalarials, antihypertensive management and nephroprotection with angiotensin-converting enzyme inhibitors and anticoagulation with enoxaparin adjusted to the renal function.
During the course of hospitalization, the patient presented progressive deterioration of the distal perfusion of fingers and toes, requiring management in the PICU. Angiotensin receptor antagonists (ARA II), topical nitroglycerin and pentoxifylline were added to management, without having obtained a response. On the contrary, there was progression of the ischemic lesion, and for this reason plasmapheresis was initiated and sympathetic blockade was performed with moderate improvement of the perfusion in the right foot. However, there was deterioration of the lesions in the left hand, marked cyanosis with areas of ischemia and necrosis in the fingers of the right hand and right foot, therefore, sildenafil and bosentan were added to management, which allowed the stabilization of the clinical picture. There was improvement in the perfusion in some areas and spontaneous exeresis of the third toe of the right foot, without requiring surgical management of the other areas of necrosis. The adequate evolution allowed the progressive reduction of bosentan. Currently, the patient is being managed with anticoagulant treatment, acetylsalicylic acid, antihypertensive and antimalarial agents and cyclophosphamide, and shows improvement in blood pressure levels and proteinuria.
During follow-up, studies were requested to rule out other factors of thrombophilia in addition to APS; among them it was obtained: homozygous mutation in the methylenetetrahydrofolate reductase gene (MTHFR) C. 677C>T (P. A222V), mutation of the prothrombin gene negative, coagulation protein C (131% within the reference range), normal factor V (FV, 85%) and factor VIII (FVIII, 51%). Therefore, is considered a prothrombotic risk factor additional to the antiphospholipid antibodies given by the presence of a mutation in the MTHFR gene.
Case 2A 16-year-old female patient, previously healthy, with no relevant family or personal antecedents was admitted due to clinical manifestations of one month of evolution characterized by respiratory symptoms, dyspnea on medium effort, chest pain and wet cough, associated with constitutional symptoms (asthenia, adinamia and fever), polyarthritis and edema in the lower limbs. During the initial assessment in the emergency department it was found a patient with arterial hypertension, tachycardia, no fever, no respiratory distress, edema in the face and lower limbs, no lesions in the oral cavity, no lymphadenopathies or megalies and no skin lesions. Leukocytopenia, normocytic normochromic anemia, negative direct Coombs test, thrombocytopenia, elevation of nitrogen compounds, proteinuria in the nephrotic range, hematuria, and schistocytes in the peripheral smear blood were evidenced in the initial paraclinical tests.
In the mediate period after admission, she presented clinical deterioration given by hemoptysis, pleural and pericardial effusion, and respiratory distress that led to hypoxemic respiratory failure. A diagnostic impression of SLE was established considering the presence of constitutional symptoms, articular, hematological (leukocytopenia, hemolytic anemia, thrombocytopenia), renal (acute renal failure, proteinuria, hematuria), and immunological (anti-DNA titer 1/640, ANA titer 1/320 homogeneous pattern, positive anti-La and anti-Ro and hypocomplementemia) commitment, pulmonary serositis (diffuse alveolar hemorrhage), thrombotic microangiopathy (schistocytosis, hemolytic anemia, negative Coombs test, elevated LDH, hyperbilirubinemia at the expense of indirect bilirubin, consumed haptoglobin and thrombocytopenia).
The patient was transferred to the PICU, where ventilatory, hemodynamic and renal support (hemodialysis) was initiated and, for her base disease, methylprednisolone pulses associated with intravenous cyclophosphamide (2 doses) were started. After the administration of the second dose, leukocytopenia and severe thrombocytopenia were evidenced and it was decided to start immunoglobulin due to refractoriness, of which she received 5 doses, without achieving control of pulmonary bleeding, for this reason, rituximab was prescribed, with adequate tolerance and control of the bleeding initially. Then she presented deterioration of the symptoms and severe thrombocytopenia and therefore, plasmapheresis was initiated for 5 sessions.
In addition, and taking into account the manifestation of thrombotic microangiopathy with ADAMTS 13 without evidence of deficiency in the enzymatic activity, it was considered an atypical hemolytic uremic syndrome and management was initiated with eculizumab, which allowed to transiently stabilize the renal function and the platelet count.
During the course of the hospitalization, a cytomegalovirus infection was documented, and for this reason the administration of the second dose of rituximab was postponed until 60 days after the first one. With the interventions performed there was partial control of the bleeding and the thrombocytopenia, improvement of renal function, with frequent exacerbations. The study was extended and associated coagulation alterations (cyanocobalamin 341pcg/mcl (reference value [RV] 189–883pcg/mcl), inhibitor of FIX 0BU, von Willebrand factor (vWF, 203%; RV 50–160%), inhibitor of FVIII 0BU, ristocetin cofactor 107.3% (RV 50–150%), FVII (111%) were ruled out. However, due to the refractoriness of the pulmonary hemorrhage with life-threatening bleeding, substitution with intravenous FVIIa was started, with initial clinical improvement.
Three months after admission, due to her torpid evolution and deterioration of the clinical status, it was performed a lung biopsy which showed widening of the interstitium due to edema, vascular congestion, foci of capilaritis and hemosiderosis. Likewise, it was performed a bone marrow aspirate, which evidenced hemophagocytes, without findings suggestive of malignancy. In addition to the histological findings, hyperferritinemia (3982ng/ml), hypertriglyceridemia and anemia were documented, and therefore it was considered a new scenario in the patient, consisting in macrophage activation syndrome (MAS), of possible multifactorial etiology. Management was initiated according to the treatment guidelines of the International Histiocyte Society of 2004 with dexamethasone, cyclosporine and etoposide, without obtaining adequate clinical response, on the contrary, with progressive clinical deterioration. Despite immunosuppressive management and daily replacement of FVIIa, the patient presented an episode of massive pulmonary bleeding which led her to death after 3 months of hospitalization.
Case 3An 8-year-old female patient who was admitted to the emergency department of the institution due to a clinical picture that began 12 months prior to the consultation, consisting of intermittent edema in the hands, morning stiffness and pain of inflammatory characteristics, which worsened 2 months prior to her admission. She presented ecchymosis in lower limbs and gingival bleeding associated and of the same time of evolution. During the initial assessment, the physical examination revealed malar erythema, epistaxis, polyarthritis (bilateral wrists, edema in the proximal interphalangeal joint of the 4th finger of the right hand and in the 2nd, 3rd and 4th fingers of the left hand), perimaleolar ecchymosis in the neck of the right foot and in the ipsilateral knee, as well as periarthritis of the right ankle, with no evidence of adenomegalies or visceromegalies. In the initial paraclinical tests, it was observed an hemogram without cytopenias, elevated acute phase reactants (ESR and CRP), renal and hepatic function within normal parameters, urinalysis with adequate urinary acidification and concentration without proteinuria, microscopic hematuria, hypocomplementemia of C3 and C4, prolonged coagulation times (PT 22.3/14.8; aPTT 59.5/29.9). The values of the autoimmunity profile were positive ANA (titer 1/2560), positive anti-dsDNA (titer 1/160), and negative extractable nuclear antigen antibodies. SLE was considered as a diagnostic impression taking into account the cutaneous, articular, renal (hematuria) and immunological (ANA, anti-DNA and hypocomplementemia) commitment. Due to the presence of prolonged coagulation times and evidence of active bleeding, a possible acquired coagulation disorder was also considered. The patient was hospitalized in order to complement the studies and initiate medical management with methylprednisolone pulses for 3 days and antimalarial medication.
During hospitalization, she had a satisfactory clinical evolution, with partial improvement in joint commitment and no evidence of new bleeding. In the paraclinical tests obtained during hospitalization, presumptive positive lupus anticoagulant, normal LDH, elevated reticulocytes, peripheral blood smear with anisocytosis without other alterations and negative direct Coombs test were evident; chest X-ray without evidence of serositis and normal abdominal ultrasound. Azathioprine was added to management and the patient was discharged with outpatient control and evaluation of the report of quantification of coagulation factors, which was still pending at that date.
In the follow-up she presented a satisfactory evolution, with control paraclinical tests with negative anti-cardiolipin antibodies, decreased FVIII (17.8%; RV 50–150%) and FIX (3.8%; RV 50–150%) levels, normal protein S, Leiden V factor, protein C and coagulation times. Hematology considered it an acquired hemophilia and, due to the adequate initial response to immunosuppressive management, it was decided to continue with the same management established. During the outpatient controls, it was observed a progressive improvement in the levels of coagulation factors: FVIII (19%) and FIX (24%), until normality was reached in the last control (FVIII: 169% and FIX: 95%), and for this reason it was considered an acquired hemophilia in remission.
Literature reviewIn the literature are reported alterations in the coagulation system in patients with SLE, as a consequence of the deficit of coagulation factors secondary to the presence of inhibitory antibodies or prothrombotic antibodies. In the 3 cases mentioned above different spectra of the disease are presented. In the first case, massive pulmonary hemorrhage; in the second case, thrombophilia due to the presence of antiphospholipid antibodies associated with the homozygous mutation in the MTHFR gene and, in the third case, a patient with acquired hemophilia. Taking into account that the patients had manifestations related to alterations in coagulation, the physiological mechanisms of coagulation will be described below, in order to understand the pathological mechanisms in each patient.
Hemostasis is the mechanism that allows to protect the body from blood loss due to any injury that occurs in the blood vessels. Its objective is to activate different physiological processes that lead to (1) the formation of the hemostatic thrombus, (2) the repair of the damage and (3) the dissolution of the clot, which is clinically manifested with the control of the hemorrhage.7,8
The coagulation cascade, which comprised a classical pathway with 2 activation pathways (intrinsic and extrinsic) that converged in a common pathway was described some decades ago.9 At present it is known that hemostasis is not possible without the cells that express the tissue factor: platelets, endothelial cells, monocytes and fibroblasts, as well as other procoagulant and anticoagulant substances.8,9
The current model of coagulation is a cell-based model. This model is triggered in the membrane of the cells that express the tissue factor and is divided into 3 phases.8,9
- 1)
Initiation phase: when the vasculature is injured and the endothelial cells are exposed, the release of inactive tissue factor on the cell surface in small quantities is favored. This factor binds to FVII (FVII) which acts as cofactor and induces its activation (FVIIa). In this way is constituted the complex tissue factor/FVIIa, which directly activates the factor X (FXa) and indirectly the factor IX (FIXa). This allows FXa to bind to the activated FV (FVa) to form a prothrombinase complex on the surface and convert prothrombin to thrombin, but in amounts that are insufficient for the formation of fibrin.7–9
- 2)
Amplification phase: the thrombin previously accumulated activates the platelets adhered by a specific receptor (glycoprotein Ia/IIa) and the vWF. In this way the thrombin activates the FV, amplifying the activity of prothrombinase and activating the FVIII (FVIIIa), which in turn acts as cofactor of FIXa to maintain the generation of the FXa. In this phase, the platelet has on its surface activated factors along with the vWF and it takes place the activation of the natural anticoagulants: TFPI (inhibitor of the TF/FVIIa complex), antithrombin and protein C, which are important in the regulation of the proagulant.7–9
- 3)
Propagation phase: on the platelet surface FXIa activates FIX. The FIXa binds to FVIIIa, establishes the tenase complex (FIXa+FVIIIa+Ca2+), catalyzes the activation of FX and increases the conformation of the FXa/FVa+Ca2+ complex. This complex catalyzes the conversion of thrombin for the formation of fibrin. The fibrin activates the FXIII (fibrin stabilizing factor), which forms covalent bonds between fibrin chains for the formation and stabilization of the clot.7–9
In SLE, thrombotic manifestations have a prevalence of 10–50%, depending on the risk factors present in each patient.7 They are mainly related to the positivity of antiphospholipid antibodies (aPL). The prevalence of lupus anticoagulant is 20–30%, of beta 2 glycoprotein 40% and of anticardiolipin antibodies 42%.9 50–80% of SLE patients with aPL meet classification criteria for APS, with increased morbidity, since it increases the risk of thrombosis 20–30-fold more than in patients without aPL.3 Cervera et al. observed, in their cohort of 1000 patients with SLE, that 9.2% of the patients had an antecedent of thrombosis during the inclusion in the study and that 1.8% of the patients included died as a consequence of thrombotic episodes, which were associated with the presence of aPL.10 The risk factors associated with thrombosis will be described below and the APS will be discussed in more detail.
High levels of homocysteine are considered as an independent risk factor for atherosclerosis, arterial and venous thrombosis.11 The hyperhomocysteinemia may be secondary to deficiency of vitamin B12, vitamin B6, folic acid, chronic renal failure, hypothyroidism or mutations in the MTHFR gene.11,12 Sallai et al. described that approximately 15% of their cohort of 105 patients with SLE had elevated homocysteine levels and that 27.3% of patients with hyperhomocysteinemia developed thrombosis compared with 16.9% who did not have thrombosis.13
MTHFR is an enzyme involved in the metabolism of homocysteine, a sulfurized amino acid product of the metabolism of methionine that comes from dietary proteins. The MTHFR gene is located on chromosome 1p36.2 and the C677T mutation consists in the substitution of a cytosine by a thymine in the 677 nucleotide, which originates the substitution of an alanine by a valine at the position 223. This change generates a thermolabile variant of MTHFR that is characterized by a decrease of 50% in its activity at 37°C, which conditions the presence of high levels of homocysteine in blood that are related to the predisposition to thrombosis.12,14,15 Among the mechanisms described by which hyperhomocysteinemia favors thrombotic episodes, are the increase in the proliferation of muscle cells, the inhibition of the synthesis of DNA of endothelial cells, the increase of the vasoconstrictor response, the reduction of the expression of thrombomodulin, the increased expression of the tissue factor, the inhibition of the expression of heparansulfate and the release of nitric oxide and prostacyclines, and the reduction of the binding of the tissue plasminogen activator to its endothelial receptor.12,13
The American Heart Association (AHA) considers that the results of the studies related to the presence of mutations in the MTHFR gene have been contradictory and that its clinical importance in the risk of thrombotic events is uncertain.15 However, some authors consider it as a risk factor additional to the presence of aPL in the development of thrombotic phenomena, as observed in the patient of case 1.16–18 Giannelou et al. identified in a cohort of 150 patients with SLE that both hyperhomocysteinemia (OR 5.8; 95% CI: 1.0–35.8) and the MTHFR 677TT genotype (OR 5.2; 95% CI: 1.1–24.0) acted as independent factors for the formation of atherosclerotic plaque. In addition, that the MTHFR genotype conferred an increased risk of thickening of the arterial wall (OR 4.9, 95% CI: 1.2–20.6) without reproducing this finding with hyperhomocysteinemia. These findings indicate that genetic influences such as the MTHFR 677TT variant increase the burden of atherosclerotic disease that characterizes SLE.19 At present there are no studies that indicate the frequency or the risk of thrombosis in patients with jSLE related to the presence of mutations in this gene.
Acquired deficits of protein S, protein C or antithrombin (AT III) are infrequent. Nevertheless, their presence favors the risk of thrombosis. Acquired protein C deficit has been reported in up to 50% of patients with lupus anticoagulant.9,20 In addition to this, in patients with nephrotic syndrome the acquired deficit of AT III can frequently be found.8 In conclusion, congenital defects that condition thrombophilias are not increased in patients with SLE, but, as previously described, their presence is associated with an increased risk of thrombotic events.3,19,20
Antiphospholipid syndromeAPS is a systemic autoimmune disease characterized by the presence of thromboembolic events, morbidity in pregnancy and high levels of aPL. 50% of patients have an isolated disease, that is, not associated with another autoimmune condition. However, it has been described that up to 50% have the APS associated with another autoimmune disease: between 40 and 80%, associated with SLE.21–23
The aPLs are antibodies that can be of the 3 idiotypes (IgG, IgM or IgA), which are directed against the protein complexes (beta 2 glycoprotein, prothrombin, annexin V, protein C, protein S) or the phospholipids of cell membranes (cardiolipin, phosphatidylserine, phosphatidylethanolamine, phosphatidylinositol). It has been described their main interaction with beta 2 glycoprotein and annexin V.21–23
The effects they generate on cells are related to platelet activation and aggregation, monocyte activation, release of proinflammatory cytokines such as IL1, IL6 and IL8 and increased expression of adhesion molecules (ICAM, VCAM and E selectin). In the endothelium, they induce the expression of the tissue factor and adhesion molecules and their effect on coagulation is related to the formation of the fibrin clot, and decreased fibrinolysis by inhibiting thrombin, protein C, plasminogen and plasmin. The effects described lead to increased tissue injury, which favors the development of thrombotic manifestations.21,22
Avcin et al. published a multicentric cohort with 121 patients with APS of onset in pediatric age. 49.5% of patients had no other autoimmune disease and 38% of patients had associated jSLE. Thrombotic manifestations were observed in 38% of patients: venous thrombosis was the most frequent (26%) and less frequently, arterial thrombosis (6%), and non-thrombotic manifestations in 40% of patients.
Hematologic involvement was described in 38%, mainly related to hemolytic anemia with positive Coombs test, mild to moderate thrombocytopenia, Evans syndrome (thrombocytopenia associated with immune hemolytic anemia) and leukocytopenia. Dermatological manifestations were reported in 18% of patients, related to livedo reticularis, Raynaud's phenomenon, cutaneous ulcers and chronic urticaria and 16% developed non-thrombotic neurological manifestations (migraine headache, movement disorders, epilepsy and affective disorders with lower frequency).24 These data are consistent with what was described by other authors.25,26
The Sapporo classification criteria to establish the diagnosis of APS have been validated in the adult population, including the morbidity in pregnancy as a clinical criterion. However, it is not considered as a classification criterion for the pediatric population.21 On the other hand, in the recommendations recently published in the EULAR guidelines is described the need to validate some classification criteria in the pediatric population in which non-thrombotic manifestations are included, since, as previously described, they are frequent clinical manifestations in this age group.25 (Table 1).22
Revised Sapporo classification criteria for APS.
| Revised Sapporo criteria in pediatric patients | |
|---|---|
| Clinical criteria | Vascular thrombosis→One of more episodes of small vessel, arterial or venous thrombosis confirmed by imaging or pathology (presence of thrombosis with evidence of inflammation in the vessel wall) |
| Laboratory criteria | Lupus anticoagulant→(+) in 2 or more occasions, difference of 12 weeks |
| IgG/IgM anticardiolipin antibodies→In serum or plasma in medium or high titers on 2 or more occasions, difference 12 weeks (ELISA) | |
| IgG/IgM beta 2 glycoprotein antibodies→In serum or plasma in high titers in 2 or more occasions, difference of 12 weeks (ELISA). | |
APS is established with a clinical and a paraclinical parameter.
Source: Taken from Petty et al.22
Although the association of aPL with thrombotic episodes has traditionally been described, these antibodies may also be directed against prothrombin and accelerate its clearance by the reticuloendothelial system, thus being associated with hemorrhagic diathesis.25
The finding of aPL is essential to establish the diagnosis. The lupus anticoagulant is more specific for APS, while anticardiolipin antibodies are more sensitive. The specificity of the latter can be increased in the presence of high titers of IgG idiotype of anti-cardiolipin antibodies.26 Although positivity is considered as a predictor of thrombosis risk in patients with SLE or APS, a definitive association between the clinical manifestations and a specific aPL has not been described.26,27 Driest et al. published a study that included 979 patients with thrombotic manifestations in jSLE, in which they found that the positivity of the aPLs was statistically significant (p=0.0052). These findings led them to define the aPLs as a risk factor for thrombosis in patients with jSLE.18
Groot et al. published the recommendations for management in patients with juvenile-onset APS. In patients with SLE and in the presence of aPL, the addition of an antiplatelet agent, ideally acetylsalicylic acid, should be considered for primary prevention. However, it should also be kept in mind that antimalarials have an antiaggregant effect. The ideal recommendation is the concomitant use of acetylsalicylic acid and an antimalarial agent. In the presence of venous thrombotic episodes and persistently positive aPLs, long-term anticoagulant therapy is indicated with an INR therapeutic target between 2 and 3. In the case of arterial thrombotic event and persistently positive aPLs, long term anticoagulant therapy or combined anticoagulant and antiplatelet therapy are indicated and an ideal INR between 2 and 3. Patients with catastrophic antiphospholipid syndrome not only require anticoagulant therapy, but the immediate use of high doses of glucocorticoids, plasmapheresis is also indicated and, in refractory cases, gammaglobulin, rituximab or other immunosuppressant should be added.28
Hemorrhagic manifestations and systemic lupus erythematosusReports of cases related to different hemorrhagic manifestations in patients with SLE have been published in the literature. In our case, patient number 2 presented alveolar hemorrhage and patient number 3 had acquired hemophilia.
Alveolar hemorrhage and systemic lupus erythematosusAlveolar hemorrhage in patients with SLE is a rare and potentially fatal clinical manifestation.29 The pathophysiological mechanisms are not completely defined. The disruption of the wall of the blood vessels by an immune-mediated pathway, the deposit of immune complexes and complement, pulmonary infections and coagulopathies have been implied.29–31
There are few case reports that have allowed to estimate an approximate frequency between 0.2 and 5.4% in patients with SLE, and it occurs more frequently in juvenile population than in adults and in the female gender.29,32 Badsha et al. described that it usually occurs in an average time of 3 years after the onset of the SLE.32 However, other authors describe that in 30% of cases alveolar hemorrhage is the first manifestation of the disease.29
The clinical manifestations include sudden onset dyspnea, respiratory distress that progresses to hypoxemic respiratory failure, cough and hemoptysis and, at the paraclinical level, a decrease in hemoglobin levels and radiological evidence of diffuse alveolar or interstitial infiltrates are observed.29–32 The mortality rate recorded in the literature is between 30 and 90% of patients.29
Araujo et al. published a retrospective study that included 13 patients with jSLE and alveolar hemorrhage. The patients included had a mean age at the onset of SLE of 12.7±4.2 years and a mean age at the onset of the alveolar hemorrhage of 15.3±2.7 years and 77% of patients were women. The reported clinical manifestations were hemoptysis, dyspnea, cough and hypoxemia in 100% of patients, a significant decrease in hemoglobin levels in 36.4% (mean decrease of 2.9±0.9g/dl) and a mortality rate of 69% of patients (all deaths were secondary to respiratory failure triggered by alveolar hemorrhage, 50% had associated infections and one patient died as a consequence of MAS).29 These findings are consistent with what was described in the case of patient number 2, who presented a cytomegalovirus infection that delayed the continuity of immunosuppressive management, with a poor response to the treatment established for the alveolar bleeding and the development of MAS that eventually led her to death.
The indicated pharmacological treatment includes high doses of steroids, cytotoxic agents, intravenous immunoglobulin and plasmapheresis.29,32 The intravenous or local administration of FVIIa is indicated in refractory cases.31 Early recognition improves the prognosis and the possibility of response to treatment.29–32
Acquired hemophilia and systemic lupus erythematosusThe presence of hemorrhagic manifestations secondary to circulating autoantibodies (inhibitors) that partially or completely neutralize the activity or accelerate the destruction of specific coagulation factors define the acquired hemophilia.33–37 It is estimated that approximately 16% of registered cases correspond to diseases of autoimmune origin, of which SLE is the most frequent.34
Inhibitors directed against FVIII are the most frequently found, hence the name acquired hemophilia A, but inhibitors of any other coagulation factor can be identified.30,35
The reported incidence is estimated at approximately 0.2–1 per million patients/year. In 50% of cases it is secondary to conditions such as postpartum, malignancy and autoimmune diseases, where SLE, rheumatoid arthritis and temporal arteritis stand out.34–36 Some authors have reported that the presence of inhibitors in SLE is found in approximately 5.6% (10 of 215 patients). However, the exact incidence is unknown, because it is often an underdiagnosed condition.34–36 The presence of acquired inhibitors is found more frequently in adult patients. In children under 16 years of age, an incidence of 0.045 per million patients is estimated.34
The main clinical manifestation is bleeding (described in 89% of patients), which can be spontaneous or occur after trauma, surgery or postpartum and its severity is variable. The hemorrhagic manifestations reported are epistaxis, gingival bleeding, ecchymosis, petechiae and even severe and potentially fatal hemorrhages in the respiratory tract, the gastrointestinal tract and the central nervous system. Between 5 and 10% of the cases described occur with severe bleeding, and for this reason it is considered a condition with a high risk of morbidity and mortality.34,35 Unlike congenital hemophilia, hemarthrosis is infrequent.34 On the contrary, a small proportion of patients do not present hemorrhagic manifestations and in these cases the prolongation of the coagulation times (aPTT or PT) is the only alteration suggestive of the disease.34,38,39
It is important to suspect the diagnosis in patients with hemorrhagic manifestations, without personal or family history relevant for hemorrhagic conditions and with the presence of abnormal coagulation times. When performing coagulation tests it is possible to find several scenarios: (a) An abnormal aPTT with normal PT and, when performing the corrected aPTT test, in which the plasma of the patient is mixed with normal plasma, if the aPTT is corrected indicates a deficiency of factors in the intrinsic coagulation pathway (XII, XI, IX, or VIII), but in those patients in whom it persists prolonged indicates the presence of inhibitors of any of the coagulation factors or the presence of antiphospholipid antibodies. (b) In abnormal PT and aPTT with a mixing test that does not correct, the presence of inhibitors directed against FV, FII and FX should be suspected.35–40
Once the diagnosis is suspected, a quantitative measurement of inhibitors (in the case of acquired hemophilia A, specific inhibitors directed against FVIII) should be performed. In addition, it is important to keep in mind that the levels of the inhibitors are not directly related with the severity of bleeding or with mortality.34 It is also possible to find low levels of FVIII, and other tests of hemostasis such as platelet count are usually normal.35 These alterations are consistent with the findings described in the patient of clinical case number 3 (prolonged PT and aPTT associated with decreased levels of FVIII and FIX), which allowed to establish the diagnosis of acquired hemophilia.
The inhibitors are quantified using the «Bethesda assay» in which normal plasma (as a source of FVIII or FIX) is mixed with undiluted plasma of the patient and this mixture is incubated during 2h at 37°C. A Bethesda unit is defined as the amount that destroys half of the factor in that corrected sample.38 The alloantibodies are polyclonal of IgG1 and IgG4 types, that are produced by loss of immune tolerance that regulates the response to FVIII.34,35
The treatment consists in controlling bleeding and eliminating the inhibitors. For the control of bleeding, the use of human or porcine FVIII, activated prothrombin concentrate (anti-inhibitor coagulant complex) or non-human recombinant FVIIa in case of severe bleeding is recommended. In patients with mild bleeding, the application of coagulation factor concentrates is not necessary.35,37
The elimination of the inhibitors requires long-term immunosuppressive therapy with corticosteroids, cytotoxic agents such as cyclosporine and cyclophosphamide or combination therapy; intravenous immunoglobulin or plasmapheresis are also indicated depending on the severity of bleeding.35–38 The current recommended regimen as first line to effectively eliminate the inhibitors is prednisolone associated with cyclophosphamide, with which an adequate response is described in 60–70% of patients.35–40 The use of biological drugs such as rituximab is indicated as second line treatment, and cyclosporine, mycophenolate mofetil, azathioprine and vincristine are recommended as an alternative.35–40
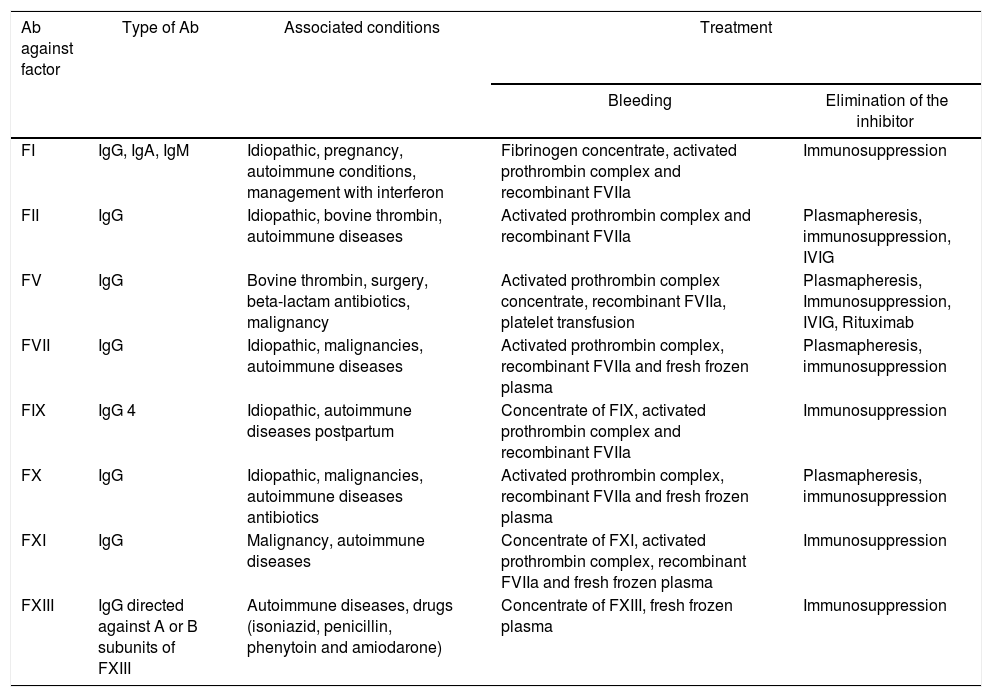
As previously mentioned, acquired inhibitors directed against other coagulation factors (I, II, V, VII, IX, X, XI and XIII) have also been described.38 The severity of the hemorrhagic manifestations is related with the involved factor and, likewise, the treatment is aimed at achieving an adequate control of bleeding and elimination of the inhibitor.40–43
The lupus anticoagulant associated hypoprothrombinemia syndrome is characterized by the presence of non-neutralizing antibodies directed against FII.39–42 It occurs in a higher proportion in the female gender and usually is manifested by severe hemorrhages that include the involvement of the central nervous system. Clinical suspicion arises in the presence of a significant prolongation of PT and aPTT, with minimal correction in the mixing test and evidence of lupus anticoagulant.40,41 Although there is no standardized management, the administration of fresh plasma or prothrombin complex concentrates associated with immunosuppressive treatment is recommended.40,42,43
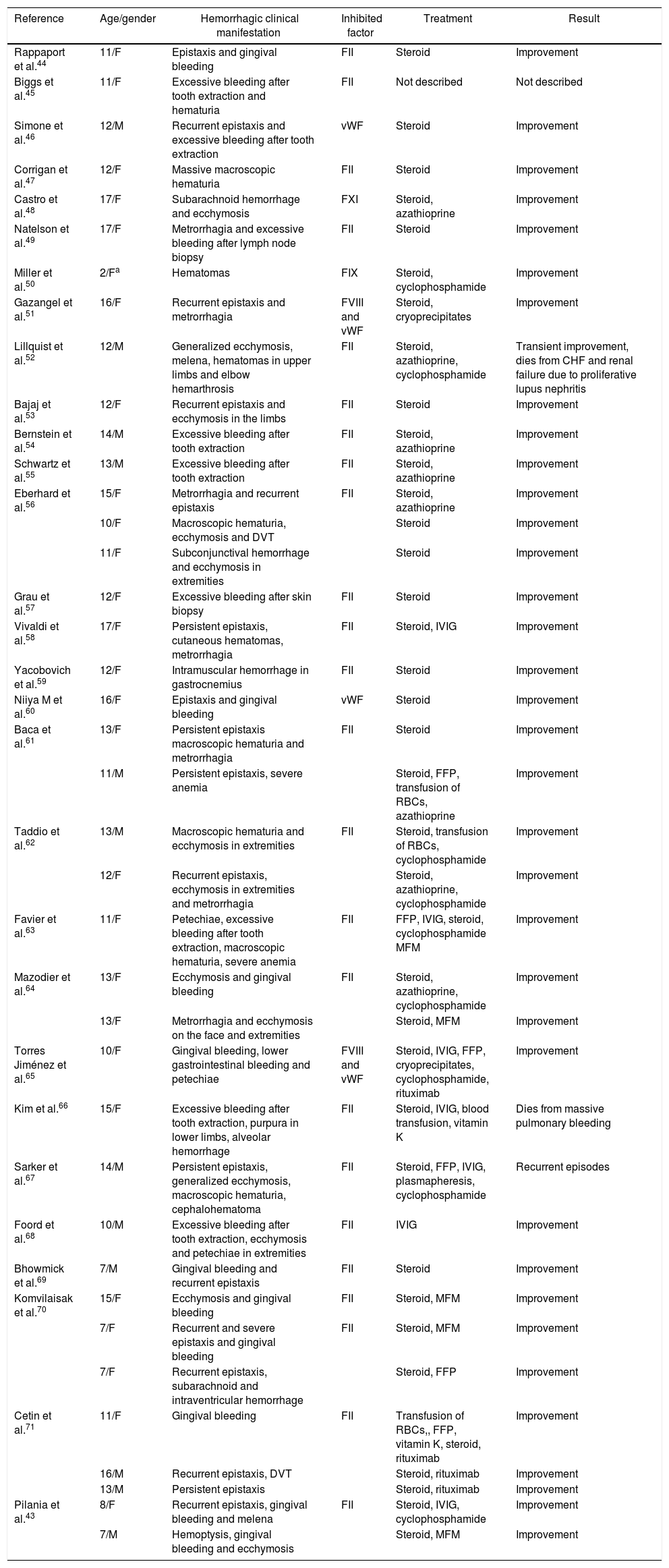
Case reports related to the presence of inhibitors and jSLE have been published in the literature. In Table 2 are described the hemorrhagic manifestations present in patients with inhibitors associated with jSLE, the compromised coagulation factor and the pharmacological requirement of the patients reported in the literature; and in Table 3 are shown the characteristics of the inhibitors for the other factors and the associated diseases. In addition, the recommended pharmacological treatment is described both for the control of bleeding and for the eradication or elimination of the inhibitor.40
Literature review of cases related with the presence of inhibitors directed against coagulation factors in jSLE.
| Reference | Age/gender | Hemorrhagic clinical manifestation | Inhibited factor | Treatment | Result |
|---|---|---|---|---|---|
| Rappaport et al.44 | 11/F | Epistaxis and gingival bleeding | FII | Steroid | Improvement |
| Biggs et al.45 | 11/F | Excessive bleeding after tooth extraction and hematuria | FII | Not described | Not described |
| Simone et al.46 | 12/M | Recurrent epistaxis and excessive bleeding after tooth extraction | vWF | Steroid | Improvement |
| Corrigan et al.47 | 12/F | Massive macroscopic hematuria | FII | Steroid | Improvement |
| Castro et al.48 | 17/F | Subarachnoid hemorrhage and ecchymosis | FXI | Steroid, azathioprine | Improvement |
| Natelson et al.49 | 17/F | Metrorrhagia and excessive bleeding after lymph node biopsy | FII | Steroid | Improvement |
| Miller et al.50 | 2/Fa | Hematomas | FIX | Steroid, cyclophosphamide | Improvement |
| Gazangel et al.51 | 16/F | Recurrent epistaxis and metrorrhagia | FVIII and vWF | Steroid, cryoprecipitates | Improvement |
| Lillquist et al.52 | 12/M | Generalized ecchymosis, melena, hematomas in upper limbs and elbow hemarthrosis | FII | Steroid, azathioprine, cyclophosphamide | Transient improvement, dies from CHF and renal failure due to proliferative lupus nephritis |
| Bajaj et al.53 | 12/F | Recurrent epistaxis and ecchymosis in the limbs | FII | Steroid | Improvement |
| Bernstein et al.54 | 14/M | Excessive bleeding after tooth extraction | FII | Steroid, azathioprine | Improvement |
| Schwartz et al.55 | 13/M | Excessive bleeding after tooth extraction | FII | Steroid, azathioprine | Improvement |
| Eberhard et al.56 | 15/F | Metrorrhagia and recurrent epistaxis | FII | Steroid, azathioprine | Improvement |
| 10/F | Macroscopic hematuria, ecchymosis and DVT | Steroid | Improvement | ||
| 11/F | Subconjunctival hemorrhage and ecchymosis in extremities | Steroid | Improvement | ||
| Grau et al.57 | 12/F | Excessive bleeding after skin biopsy | FII | Steroid | Improvement |
| Vivaldi et al.58 | 17/F | Persistent epistaxis, cutaneous hematomas, metrorrhagia | FII | Steroid, IVIG | Improvement |
| Yacobovich et al.59 | 12/F | Intramuscular hemorrhage in gastrocnemius | FII | Steroid | Improvement |
| Niiya M et al.60 | 16/F | Epistaxis and gingival bleeding | vWF | Steroid | Improvement |
| Baca et al.61 | 13/F | Persistent epistaxis macroscopic hematuria and metrorrhagia | FII | Steroid | Improvement |
| 11/M | Persistent epistaxis, severe anemia | Steroid, FFP, transfusion of RBCs, azathioprine | Improvement | ||
| Taddio et al.62 | 13/M | Macroscopic hematuria and ecchymosis in extremities | FII | Steroid, transfusion of RBCs, cyclophosphamide | Improvement |
| 12/F | Recurrent epistaxis, ecchymosis in extremities and metrorrhagia | Steroid, azathioprine, cyclophosphamide | Improvement | ||
| Favier et al.63 | 11/F | Petechiae, excessive bleeding after tooth extraction, macroscopic hematuria, severe anemia | FII | FFP, IVIG, steroid, cyclophosphamide MFM | Improvement |
| Mazodier et al.64 | 13/F | Ecchymosis and gingival bleeding | FII | Steroid, azathioprine, cyclophosphamide | Improvement |
| 13/F | Metrorrhagia and ecchymosis on the face and extremities | Steroid, MFM | Improvement | ||
| Torres Jiménez et al.65 | 10/F | Gingival bleeding, lower gastrointestinal bleeding and petechiae | FVIII and vWF | Steroid, IVIG, FFP, cryoprecipitates, cyclophosphamide, rituximab | Improvement |
| Kim et al.66 | 15/F | Excessive bleeding after tooth extraction, purpura in lower limbs, alveolar hemorrhage | FII | Steroid, IVIG, blood transfusion, vitamin K | Dies from massive pulmonary bleeding |
| Sarker et al.67 | 14/M | Persistent epistaxis, generalized ecchymosis, macroscopic hematuria, cephalohematoma | FII | Steroid, FFP, IVIG, plasmapheresis, cyclophosphamide | Recurrent episodes |
| Foord et al.68 | 10/M | Excessive bleeding after tooth extraction, ecchymosis and petechiae in extremities | FII | IVIG | Improvement |
| Bhowmick et al.69 | 7/M | Gingival bleeding and recurrent epistaxis | FII | Steroid | Improvement |
| Komvilaisak et al.70 | 15/F | Ecchymosis and gingival bleeding | FII | Steroid, MFM | Improvement |
| 7/F | Recurrent and severe epistaxis and gingival bleeding | FII | Steroid, MFM | Improvement | |
| 7/F | Recurrent epistaxis, subarachnoid and intraventricular hemorrhage | Steroid, FFP | Improvement | ||
| Cetin et al.71 | 11/F | Gingival bleeding | FII | Transfusion of RBCs,, FFP, vitamin K, steroid, rituximab | Improvement |
| 16/M | Recurrent epistaxis, DVT | Steroid, rituximab | Improvement | ||
| 13/M | Persistent epistaxis | Steroid, rituximab | Improvement | ||
| Pilania et al.43 | 8/F | Recurrent epistaxis, gingival bleeding and melena | FII | Steroid, IVIG, cyclophosphamide | Improvement |
| 7/M | Hemoptysis, gingival bleeding and ecchymosis | Steroid, MFM | Improvement |
Patient diagnosed with autoimmune disease with positive ANA, high suspicion of jSLE.
F: female gender; vWF: von Willebrand Factor; RBC: red blood cells; CHF: congestive heart failure; IVIG: intravenous immunoglobulin; M: male gender; MFM: mycophenolate mofetil; FFP: fresh frozen plasma; DVT: deep venous thrombosis.
Characteristics of acquired inhibitors of coagulation factors.
| Ab against factor | Type of Ab | Associated conditions | Treatment | |
|---|---|---|---|---|
| Bleeding | Elimination of the inhibitor | |||
| FI | IgG, IgA, IgM | Idiopathic, pregnancy, autoimmune conditions, management with interferon | Fibrinogen concentrate, activated prothrombin complex and recombinant FVIIa | Immunosuppression |
| FII | IgG | Idiopathic, bovine thrombin, autoimmune diseases | Activated prothrombin complex and recombinant FVIIa | Plasmapheresis, immunosuppression, IVIG |
| FV | IgG | Bovine thrombin, surgery, beta-lactam antibiotics, malignancy | Activated prothrombin complex concentrate, recombinant FVIIa, platelet transfusion | Plasmapheresis, Immunosuppression, IVIG, Rituximab |
| FVII | IgG | Idiopathic, malignancies, autoimmune diseases | Activated prothrombin complex, recombinant FVIIa and fresh frozen plasma | Plasmapheresis, immunosuppression |
| FIX | IgG 4 | Idiopathic, autoimmune diseases postpartum | Concentrate of FIX, activated prothrombin complex and recombinant FVIIa | Immunosuppression |
| FX | IgG | Idiopathic, malignancies, autoimmune diseases antibiotics | Activated prothrombin complex, recombinant FVIIa and fresh frozen plasma | Plasmapheresis, immunosuppression |
| FXI | IgG | Malignancy, autoimmune diseases | Concentrate of FXI, activated prothrombin complex, recombinant FVIIa and fresh frozen plasma | Immunosuppression |
| FXIII | IgG directed against A or B subunits of FXIII | Autoimmune diseases, drugs (isoniazid, penicillin, phenytoin and amiodarone) | Concentrate of FXIII, fresh frozen plasma | Immunosuppression |
Ab: antibodies; Ig: immunoglobulin; IVIG: intravenous immunoglobulin.
Source: Taken from Franchinni et al.40
Hematological alterations are common in patients with SLE. It is important to differentiate between the manifestations related to the disease and those that are secondary to the treatment. The hematological involvement can be differentiated according to whether it is related to the commitment of the cell lineages, evidenced by the presence of leukocytopenia, thrombocytopenia, anemia (hemolytic anemia, anemia of chronic disease and anemia due to iron deficiency) and alterations related to the involvement of coagulation.
Coagulation commitment is associated with thrombotic and hemorrhagic manifestations. The presence of aPL favors the presence of thrombotic manifestations, at the arterial or venous level, and non-thrombotic manifestations frequently described in patients with APS isolated or associated with SLE. The non-thrombotic manifestations include hematological findings (immune thrombocytopenia, hemolytic anemia with positive Coombs test and leukocytopenia), cutaneous involvement (Raynaud's phenomenon, livedo reticularis, skin ulcers) and neurological compromise (migraine headache, movement disorders, epilepsy and affective disorders less frequently). There are risk factors additional to the presence of aPL that favor the development of thrombotic manifestations such as the presence of elevated homocysteine levels (secondary to cyanocobalamin deficit, chronic renal failure, hypothyroidism and mutations in the MTHFR gene), acquired deficit of protein S, protein C and antithrombin.
Hemorrhagic diathesis has also been described less frequently. Alveolar hemorrhage is a rare manifestation, but with high mortality, which requires early diagnosis and aggressive treatment. Hemorrhagic manifestations are also related to the acquired deficit of coagulation factors in relation to the presence of inhibitors.
It is estimated that approximately 16% of cases of acquired inhibitors occur in the context of autoimmune diseases, with a higher proportion in patients with SLE. It is important to have a high index of suspicion in patients with hemorrhagic disorders, without a family or personal history of hemorrhages, with prolonged coagulation times that do not correct with the mixing test.
Early diagnosis and treatment reduce the morbidity and mortality related to this type of manifestations in SLE.
Conflict of interestNone of the authors declared conflict of interest.
Please cite this article as: Reina Ávila MF, Saza Mejía LM, Guarnizo Zuccardi PR, Rengifo L, Garcés Sterling SP. Manifestaciones relacionadas con alteraciones en la coagulación en lupus eritematoso sistémico de inicio juvenil. Reporte de casos y revisión de la literatura. Rev Colomb Reumatol. 2020;27:190–201.
