



Oncogenic osteomalacia is a paraneoplastic metabolic syndrome characterised by a low phosphates in the blood due to renal phosphate losses with inadequately normal or low vitamin D levels. This disorder is associated with the release of tumour cell-secreted phosphaturic factor, most notably fibroblast growth factor 23 (FGF-23). The neoplasms related to oncogenic osteomalacia are usually small tumours of mesenchymal lineage, and they may be difficult to locate in the physical examination in some cases, due to their size and inaccessible location. The case is presented of a patient who developed vertebral and hip fractures due to oncogenic osteomalacia associated with a phosphaturic mesenchymal tumour of the deep fat tissue in the sole of the foot. This was finally diagnosed after 3 years of the onset of symptoms after being located by bone scintigraphy with Indium 111-labelled pentetreotide and magnetic resonance imaging.
La osteomalacia oncogénica es un síndrome metabólico paraneoplásico caracterizado por hipofosfatemia debida a la pérdida renal de fosfato con niveles inadecuadamente normales o bajos de vitamina D. Este trastorno está asociado con la liberación de factores fosfatúricos por células tumorales, especialmente el factor de crecimiento fibrolástico 23 (FGF23). Las neoplasias relacionadas con la osteomalacia oncogénica suelen ser tumores pequeños de linaje mesenquimatoso y pueden ser difíciles de localizar en algunos casos debido a su tamaño y ubicación poco accesible al examen físico. Presentamos a un paciente que desarrolló fracturas vertebrales y de cadera debido a osteomalacia oncogénica asociada con un tumor mesenquimatoso fosfatúrico del tejido graso profundo de la planta del pie, que finalmente se diagnosticó después de 3 años del inicio de los síntomas cuando el tumor pudo ser localizado por el rastreo gammagráfico óseo con pentatreótido marcado con Indio-111 y las imágenes de resonancia magnética nuclear.
Oncogenic osteomalacia (OO), also known as tumor-induced osteomalacia, is a paraneoplastic metabolic syndrome characterised by hypophosphatemia caused by phosphate kidney losses with abnormal or low levels of 1.25-hydroxyvitamin D [1.25(OH)-D]. It is associated with tumor cells release of factors previously known as “phosphatonins”, particularly the fibroblast growth factor 23 (FGF23), which behaves as a phosphaturic hormone.1 The neoplasms associated with this disorder are usually small mesenchymal linage tumors.2,3 This case discusses a patient who developed vertebral and hip fractures due to OO associated with a small phosphaturic mesenchymal tumor of the deep adipose tissue of the sole.
Case presentationThe patient is a 48-year old male from Peru, with unremarkable personal or family history. When the patient was 43 years old, he visited de doctor because of mechanical lumbar pain, irradiating to the proximal region of both lower extremities, with associated mostly proximal muscle weakness. During the physical examination the patient complaint of pain when applying pressure to the lumbar spinous processes at L4 and L5 and in the paravertebral muscles at the same levels. Muscle strength could not be assessed in the lower extremities because of pain and difficulty to walk; heel-to-toe walking was also difficult. The blood biochemistry results (including calcium level) and CBC, were within the normal ranges at that time, so no bone metabolism tests were required. The lumbar spine MRI showed a small central hernia at the level of L5-S1, a disc protrusion at L4-L5, and lumbar spinal stenosis at L3-L5; hence the symptoms were attributed to these findings.
The patient underwent L3-L4-L5-S1 arthrodesis and decompression with laminectomy. After the procedure the patient did not experience any clinical improvement, with persisting pain and weakness of the lower extremities that limited his gait and required the use of a cane. In the following two years the patient was re-operated twice due to loosening of the osteosynthesis material; during the last intervention, “poor bone quality” was identified. After curettage, the cancellous bone was soft, elastic and easy to cut with the surgical knife. The histopathological findings of the surgical specimens were unspecific and were reported as trabecular laminar bone with varying sizes of the trabecula, very scarce intertrabecular adipose tissue or replaced by serofibrinous degeneration. During this last hospital admission, the x-rays showed a significant number of grade II lumbar and dorsal vertebral crushing – according to Genant classification – and in the hips, sequelae of old basicervical type fractures with secondary varus-impacted deformity, and signs of bilateral coxarthrosis. This changes were suspicious of bone metabolism disorders, so the patient was referred to us for osteoporosis and metabolic disease screening. During the re-assessment, the patient denied any previous fragility fractures or a family history of this type of fractures. No changes were identified in dentition, the color of the sclera, or ligamentous hyperlaxity. At this moment, during the examination the patient presented pain when applying pressure over the lumbar and dorsal apophyses, and experienced limited hip mobility at 90°flexion and 20° internal and external rotation. The blood tests revealed the following values: total alkaline phosphatase (APh) 342 U/l (normal values (nv): 53-128); bone Alkaline phosphatase 290 U/l (85% of the total APh, normal values: 32-83); calcium 9.1mg/dl (nv: 8.7-10.3); Phosphorus 1.9mg/dl (nv: 2.7-4.5); 1.25(OH)-D<5pg/mL (nv: 19-48), 25(OH)-D 11ng/mL (nv: 19.1-57.6); parathormone 55.3pg/mL (nv: 12-65); osteocalcin 31.7ng/mL (nv: 15-46); C-terminal telopeptide of type I collagen 0.824ng/mL (nv: 0-0,550); and procollagen type 1 N-terminal propeptide 375.90ng/mL (nv: 12-62). The 24-h urine biochemistry showed tubular phosphate reabsorption of 69.91% (nv: 80-94), with increased phosphate excretion a of 1,440mg/24h (nv: 400-1,300) and normal excreted calciuria (60mg/24h), without any other alterations. The study was completed with tryptase and androgen levels which were found to be normal.
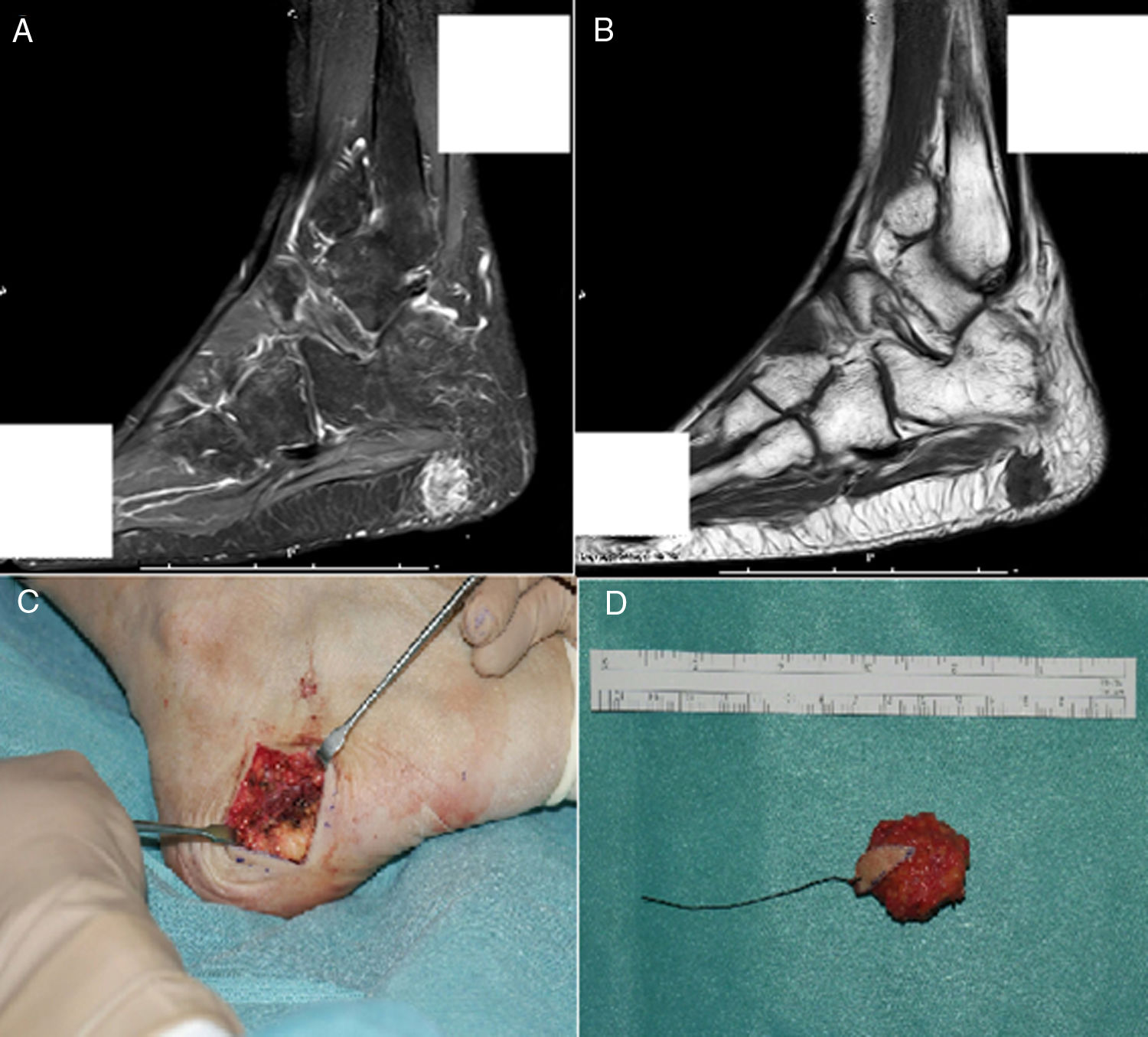
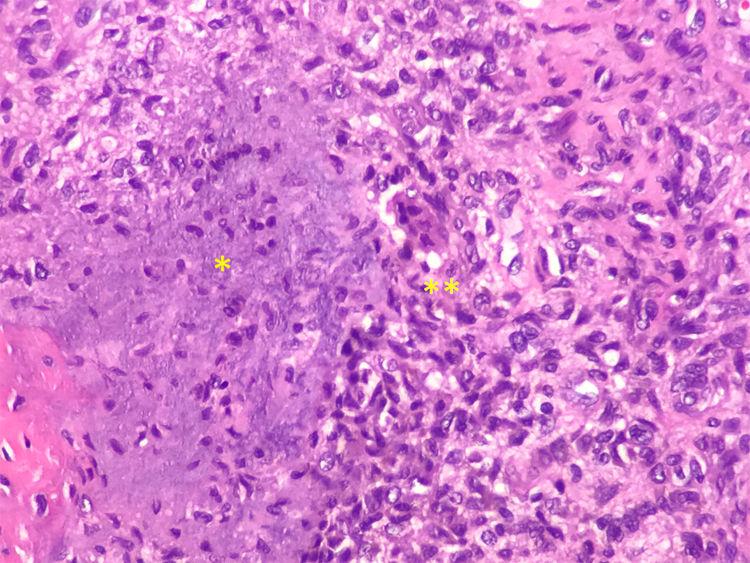
The final diagnosis was hypophosphatemic osteomalacia and phosphate therapy was initiated (4g/day) and calcitriol (0.25μg/day). After one month of therapy the phosphate blood levels were normalized and there was a mild clinical improvement in muscle pain. Considering the possibility of a tumor etiology, an Indium-111 (111In) pentetreotide bone scan was performed, showing a dotted uptake in the sole of the right foot. This finding was confirmed with MRI, identifying a solid 1.7×1.4cm tumor in the plantar hindfoot fat, with gadolinium enhancement (Fig. 1A and B). The encapsulated, free-margin tumor was excised (Fig. C), and the histopathology revealed a nodular growth mesenchymal proliferation, consisting of spindle cells, with no atypia or pleomorphism, well vascularized by hemangiopericytoma-like vessels, and by abundant giant cells, with no atypia and with basophil calcification foci with a very low proliferation index (Fig. 2). All of these findings were compatible with a phosphaturic mesenchymal tumor, though the FGF23 level could not be established in this case because of technical limitations. Following the procedure, the patient’s phosphorus blood levels became normal (3.1mg/dl) and was able to discontinue the phosphate supplements in less than one month. 3 years after the onset of symptoms, finally the OO diagnosis was made. The control laboratory tests 9 months later showed phosphorus levels of 3.9mg/dl), vitamin D [25-(OH)D] 27ng/mL; phosphate tubular reabsorption 91.4% and excreted phosphaturia 874mg/24h, all within normal ranges; APh decreased to 132mg/dl. A total bilateral hip replacement significantly improved the patient’s functional limitation.
 MRI showing a polylobulated lesion of approximately 17×14mm, contrast enhanced in the STIR sequence. B) Hypointense in T1, localized inside the subcutaneous fat of the posterior plantar region, with no clear dependency of the plantar fascia. C) Cross-section incision in the right lateral plantar region during surgery. D) Macroscopic aspect of the encapsulated tumor excised with free margins.")
A) MRI showing a polylobulated lesion of approximately 17×14mm, contrast enhanced in the STIR sequence. B) Hypointense in T1, localized inside the subcutaneous fat of the posterior plantar region, with no clear dependency of the plantar fascia. C) Cross-section incision in the right lateral plantar region during surgery. D) Macroscopic aspect of the encapsulated tumor excised with free margins.
 calcified matrix production (*), with osteoclast-type giant cells (**). Hematoxylin – eosin stain (20×).")
OO is a rare condition, with less than 400 cases reported in the literature to this date. The clinical manifestations are bone or muscle pain, associated with proximal muscle weakness and – like in our case – fragility fractures (vertebral, costal or femoral neck) which may be present in over 80% of the patients prior to diagnosis.2 Due to the non-specific nature of the symptoms, the mean time elapsed until finally making a diagnosis of OO is around 5 years, depending on the series, and there can be delays of up to 20 years.2,3
The suspicious diagnosis of OO is established based on the characteristic laboratory findings: hypophosphatemia with increased urinary phosphate excretion, increased alkaline phosphatase levels and low to normal levels of 1.25(OH)-D, in the presence of normal calcium. The parathormone may be normal or elevated in the context of hyperparathyroidism secondary to the vitamin D deficiency.1 All of these findings are associated with the production of FGF23 by the tumor cells, affecting the renal tubules and increasing the phosphate kidney excretion, in addition to reducing the production of 1.25(OH)-D.4 However, as a result of the analytical findings, a differential diagnosis should be made with other causes of hereditary or acquired hypophosphatemic osteomalcia,5 including toxins (alumina, arsenic), medications (IV iron, nucleotide-analogue antiretrovirals) and Fanconi syndrome,1,6,7 which were all ruled out in this patient following the study of aminoaciduria, glucosuria and arterial blood gases, which were all normal. Other hereditary conditions resulting from excessive FGF23, such as autosomal dominant hypophosphatemic osteomalacia, chromosome X-related HO or the autosomal recessive type, all present identical biochemical findings to OO,4 but the onset of the disease in adulthood, the symptoms of proximal muscle weakness and pain in the lower extremities, as well as the absence on family history or prior metabolic disorders, ruled out this possibility in our case.
Most OO-associated tumors are of a benign mesenchymal-origin, and are slow growing neoplasms, usually solitary and localized in the bones (around 30%) or in soft tissues (around 60%).2,3 They usually develop in the lower extremities (55–60%), and less often in the head and neck (23–30%) and upper limbs (around 5%).2,3 The mesenchymal tumors reported in the literature include: hemangiopericytoma, giant cells bone tumor, sclerosing hemangioma, angiolipoma, and the group of tumors currently classified as phosphaturic mesenchymal tumors, with various subtypes, including: osteoblastoma-like tumors, ossifying fibroma-like, non-ossifying fibroma-like tumors and the PMTMCT (phosphaturic mesenchymal tumor with mixed connective tissue), which is the most frequent type and represents around 70–80% of the mesenchymal OO-associated tumors.
From the histological point of view, these tumors have a polymorphic appearance, made up by a mixture of spindle cells, osteoclast-like giant cells, prominent blood vessels, cartilaginous matrix and metaplastic bone.8,9 Moreover, malignant variants have been described with a more sarcomatoid histology (greater atypia and mitotic activity). Metastases have been described in less than 5% of these cases, and usually these are lung metastases.1,8,10 Since these are usually small tumors, commonly asymptomatic and easily missed by a physical examination, the functional diagnostic techniques are particularly relevant. A full body examination is important, since they may develop in a distal segment of the extremities.11
These tumors frequently express somatostatin receptors; a bone scan with an analogue of this hormone (pentatreotide) labeled with 111In has been used for improved detection.12 Other techniques used include PET-CT with fluorodeoxyglucose, and more recently, PET/CT with Ga68-DOTA-1-NaI3-octreotide (DOTANOC). FGF23 venous sampling has also been used, for which venous determinations are performed in the 4 extremities and the level of FGF23 is determined for each limb, in order to facilitate the localization of the tumor.13,14
The identification of suspicious hypermetabolic areas requires anatomical confirmation using MRI or CT, since in some cases the uptake may be associated with fractures. If the clinical presentation is compatible, but the tumor has not been localized, the recommendation is to repeat the tests in 1–2 years.2,3
The treatment of choice in these cases is complete surgical tumor resection, which is curative, otherwise there may be local tumor recurrence.15 Following the intervention, the recovery from phosphatemia is usually observed between 5 and 10 days, and that of APh between 9 and 24 months.2 If phosphatemia fails to normalize, or if it drops again, this is indicative of local recurrence or metastasis.15 The impossibility to localize the tumor or when the tumor is not resectable, medical therapy with phosphorus supplementation should be initiated at a dose of 1−3g/day, divided into 4–6 doses, and calcitriol 15−60ng/kg /day, monitoring treatment response for the potential development of secondary hyperparathyroidism.1 Among the alternative therapies to surgery in case of unresectable tumors, radiofrequency ablation has been used.16 Some cases have been reported treated with subcutaneous octreotide,17 and the use of anti-FGF23 (KRN23) monoclonal antibodies is being studied; these have proven to be effective in patients with chromosome X-associated osteomalacia.18
ConclusionsOO is an important challenging diagnosis, since it is usually insidious and symptoms are unspecific. Although it is usually caused by benign tumors, it should be monitored in the presence of compatible biochemical imbalances, since it could result in significant morbidity, particularly in young patients, because of the development of insufficiency fractures and subsequent disability. Although determining the FGF23 levels is helpful to guide the diagnosis of OO, the patient’s clinic prevails, as well as all the steps leading to a diagnosis, including laboratory tests and imaging studies, both functional such as bone scan, and morphological such as MRI, in order to identify and accurately localize the OO-associated tumors, which are not usually detected in the physical examination because of their small size and deep plane localization, particularly in the lower extremities.
Conflict of interestsThe author have no conflict of interests to disclose.
Please cite this article as: Garrote-Corral S, Sifuentes-Giraldo WA, Reguero-Callejas ME, Vázquez-Díaz M. Osteomalacia oncogénica secundaria a tumor mesenquimal fosfatúrico del pie. Rev Colomb Reumatol. 2020;27:210–214.
