


Las disgenesias gonadales mixtas (DGM) son trastornos de la diferenciación sexual poco frecuentes, pero constituyen una causa importante de infertilidad. Presentan un cariotipo en mosaico con fórmula mos 45,X/46,XY y pueden dar lugar a gran variedad de fenotipos, encontrando desde diferentes grados de ambigüedad sexual en recién nacidos, hasta fenotipos masculinos normales, fenotipos femeninos normales o fenotipos del síndrome de Turner (ST).
Se presenta el caso de una paciente diagnosticada de ST desde la pubertad a quien no se le detectó la presencia de fragmentos de cromosoma Y. Teniendo en cuenta que las pacientes diagnosticadas de ST con expresión de cromosoma Y (completo o parcial) tienen mayor riesgo de desarrollar gonadoblastoma, es importante resaltar la importancia de diagnosticar la presencia de cromosoma Y, recomendando incluso realizar de forma sistemática técnicas que aumenten la sensibilidad para detectarlo aunque no se haya detectado en el cariotipo.
Mixed gonadal dysgenesis is a group of rare disorders of sexual differentiation and is a major cause of infertility. They show a mosaic karyotype 45,X/46,XY and can give rise to a great variety of phenotypes, finding from different degrees of sexual ambiguity in newborns, up to normal male phenotypes, normal female phenotypes or Turner syndrome (TS) phenotypes.
The case is presented of a patient diagnosed with TS from puberty and in whom the presence of fragments of Y chromosome was not detected. Given that patients with a diagnosis of TS with Y chromosome expression (full or partial) are at increased risk of developing gonadoblastoma, it is important to emphasise the importance of diagnosing the presence of the Y chromosome, and even recommending systematically performing techniques that increase the sensitivity in order to detect it, even though it has not been detected in the karyotype.
La disgenesia gonadal (DG) es un trastorno del desarrollo embrionario que impide la maduración completa del tejido gonadal en su diferenciación hacia testículo u ovario. Suele diagnosticarse al nacimiento, por la presencia evidente de ambigüedad de genitales externos o durante la infancia o adolescencia temprana, por signos de pubertad precoz o, por el contrario, retrasada.
Desde el año 2006 estas DG1 se clasifican en 1) anomalías de la diferenciación sexual (ADS) 46,XX; 2) ADS 46 XY, y 3) ADS por alteraciones cromosómicas, donde se encuentran las disgenesias gonadales mixtas (DGM) que presentan un cariotipo en mosaico mos 45,X/46,XY que puede dar lugar a gran variedad de fenotipos, desde diferentes grados de ambigüedad sexual en recién nacidos, hasta fenotipos masculinos normales, fenotipos femeninos normales o fenotipos del síndrome de Turner (ST).
Las pacientes diagnosticadas de ST con expresión de cromosoma Y (completo o parcial) tienen mayor riesgo de desarrollar gonadoblastoma2, estando indicada la gonadectomía profiláctica desde edades tempranas3. Por lo cual, es importante detectar la presencia de cromosoma Y en el análisis citogenético, sobre todo si encontramos en el cariotipo un fragmento cromosómico de origen desconocido. Se recomienda realizar PCR para detección de cromosoma Y aunque no se haya detectado en el cariotipo si existe cierto grado de virilización, e incluso en las últimas revisiones se recomienda realizarlo de forma sistemática a todas las pacientes diagnosticadas de ST4.
Paciente de 31 años de sexo femenino, que acude al servicio de Ginecología por esterilidad primaria solicitando consejo reproductivo, sin otros síntomas asociados.
Como antecedentes patológicos presenta: amenorrea primaria, por lo que se diagnosticó de ST mediante cariotipo del que no se dispone y se encuentra en tratamiento con estradiol más norgestrel desde los 17 años, intolerancia a la glucosa, escoliosis intervenida, nevus cutáneos e hipoacusia neurosensorial bilateral.
A la exploración presenta: desarrollo de caracteres sexuales secundarios normales, vello, mamas y genitales externos femeninos normales; peso: 69,9kg; talla: 1,43m; IMC: 34,18kg/m2. En la eco transvaginal se aprecia útero normal, anexo derecho compatible con cintilla/ovario rudimentario, y no se visualiza ovario izquierdo.
Desde la consulta de Ginecología se solicita:
- -
Hemograma y bioquímica realizada 2 meses después del cese del tratamiento estrogénico para llevar a cabo una correcta valoración de las hormonas.
- -
Estudio cardiológico para descartar malformaciones del ST.
- -
Cariotipo en sangre periférica pues no aporta ningún informe citogenético que confirme el diagnóstico.
El estudio de marcadores tumorales y hormonal se realizó mediante inmunoquimioluminiscencia (Arquitect i2000) determinándose los valores plasmáticos de AFP (alfafetoproteína), CA19.9 (antígeno carcinoembrionario 19.9), CA125 (antígeno carcinoembrionario 125), FSH (folitropina), LH (lutropina), estradiol-17-beta, TSH (hormona tiroestimulante), T4L (tiroxina 4 libre), βHCG (hormona gonadotrofina coriónica humana) y testosterona.
Para el estudio citogenético y molecular, se realizó la técnica estándar para cultivar linfocitos de sangre periférica, y las preparaciones fueron tratadas con tripsina para obtener el bandeo G.
Para el análisis microscópico se capturaron más de 20 metafases informativas con el equipo Metafer® y se clasificaron con la ayuda del programa IKAROS®.
En el análisis de hibridación in situ por fluorescencia (FISH) se utilizaron las siguientes sondas: 1) la sonda centromérica para el cromosoma X y para el cromosoma Y hibridando específicamente las secuencias de ADN satélites alfa específicas en la región del centrómero Yp11.1-q11.1 (DYZ3) Cytocell®; 2) la sonda DYZ1 (Appligene- Oncor®) que se hibrida específicamente con el ADN satélite específico para el locus DYZ1 contenido dentro de la región Yq12 del cromosoma Y; 3) la sonda Yp11.3 que determina el gen SRY (Vysis®).
Los resultados bioquímicos mostraron hipercolesterolemia y una elevación de las transaminasas, sin ningún otro hallazgo significativo. Los resultados de los marcadores tumorales se encontraron dentro de la normalidad: AFP de 6,8ng/mL (rango de normalidad entre 0-8ng/mL), CA19.9 de 2,6UI/mL (rango de normalidad entre 0-40UI/mL) y CA125 de 4,2UI/mL (rango de normalidad entre 0-35UI/mL). Los resultados del estudio hormonal manifestaron un eje tiroideo normal, LH de 15mUI/mL (rangos de normalidad de LH en mUI/mL en menopausia: 10,4-64,6), FSH de 36,4mUI/mL (rangos de normalidad de FSH en mUI/mL en menopausia: 10,4-64,6), estradiol-17-beta <10pg/mL (valores de normalidad del estradiol-17-beta en menopausia: <10-28pg/mL), βHCG <1,2mUI/mL (valores de normalidad: 0-4mUI/mL) y testosterona de 0,2ng/mL (valores de normalidad: 0,3-1,3ng/mL).
En el ecocardiograma-doppler no se encontraron hallazgos patológicos.
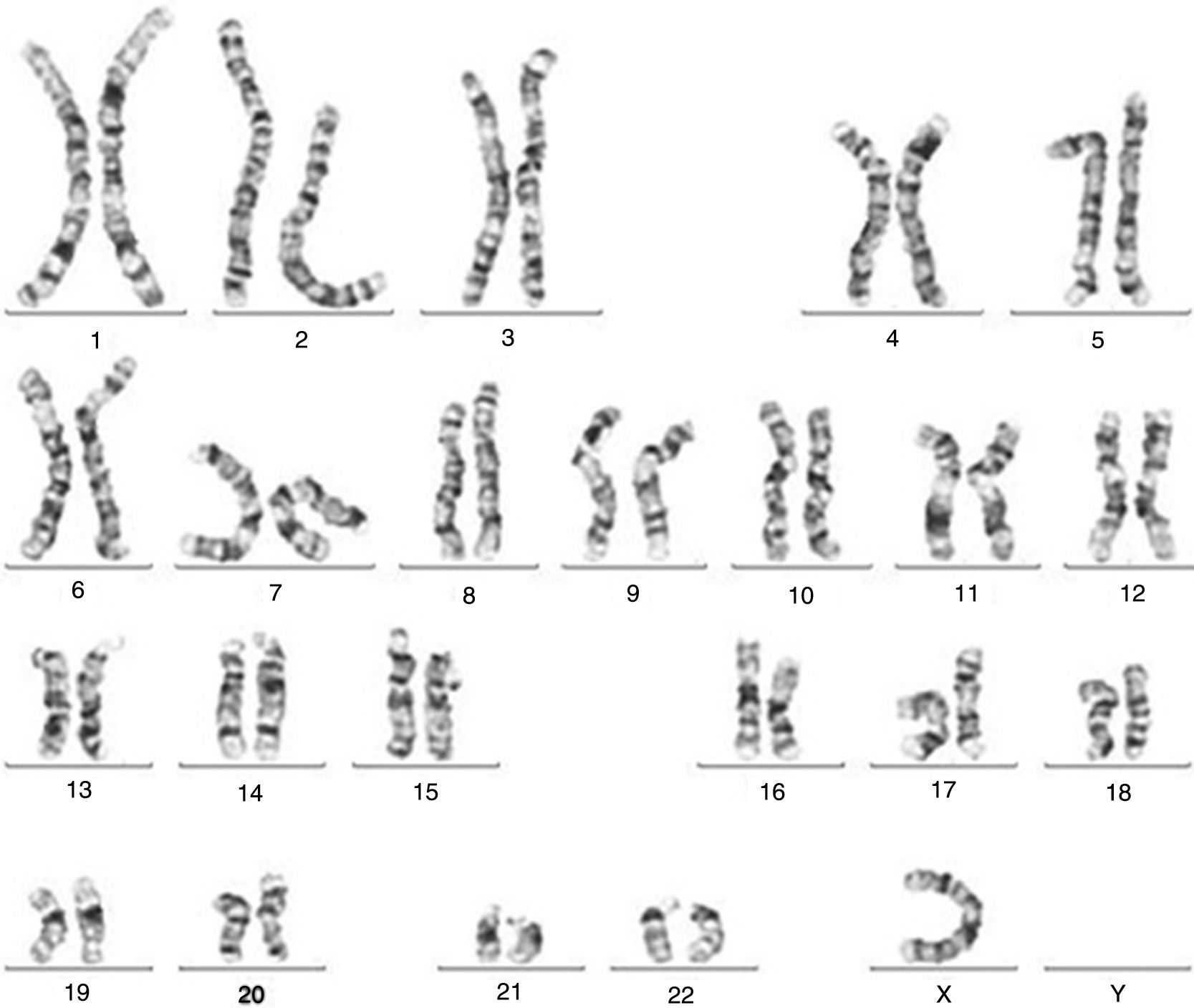
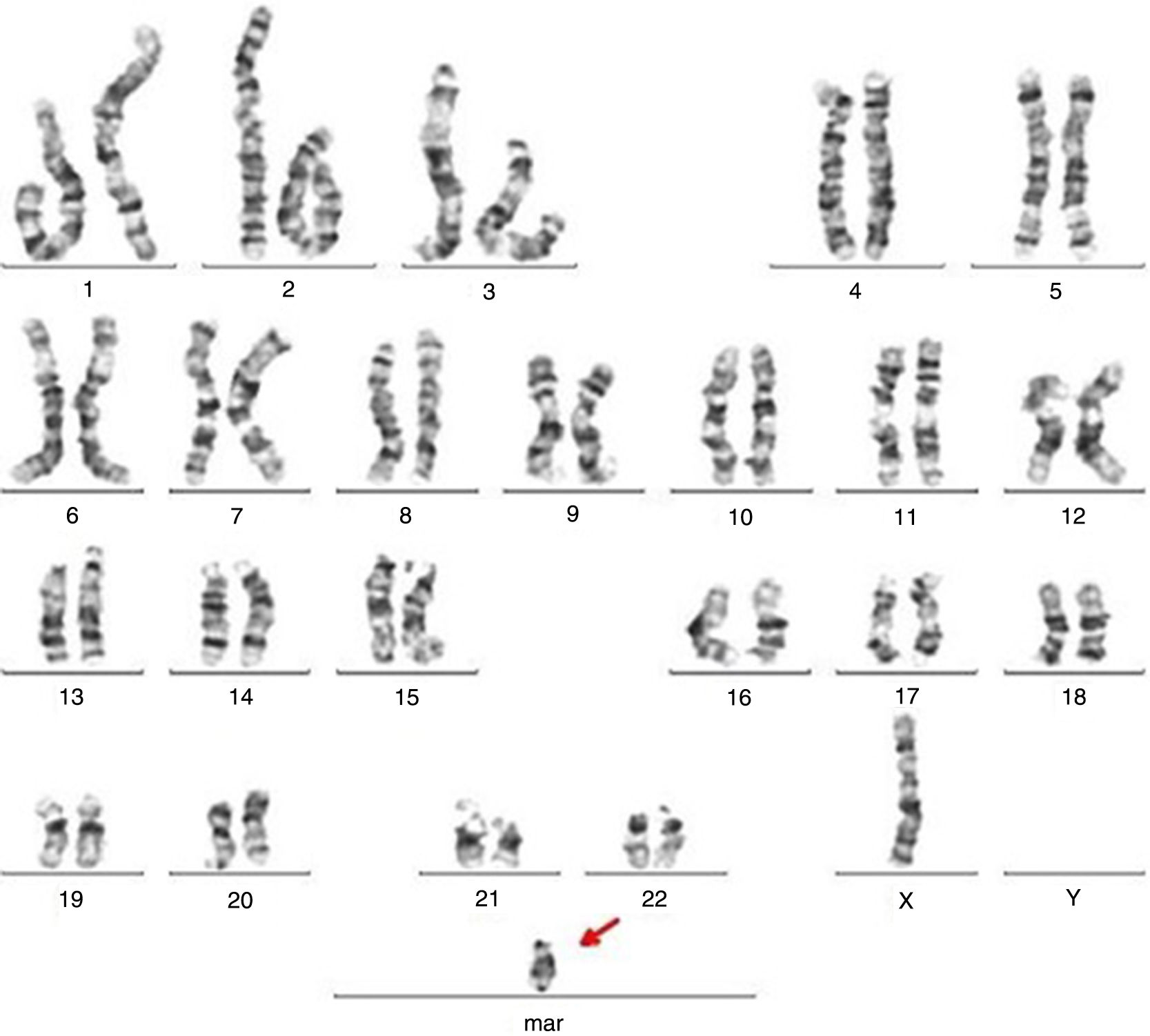
En el estudio citogenético se obtuvieron dos líneas celulares (figs. 1 y 2): una mayoritaria con fórmula cromosómica 45,XO y otra línea celular que mostró la presencia de un cromosoma marcador cuya estructura en un principio no permitió clasificarlo dentro de los grupos definidos en el estándar, siendo su fórmula cromosómica 46,X+(mar). Siguiendo los criterios International System for Human Cytogenetic Nomenclature 2016 (ISCN 2016) se informó mos 45,XO [30] /46,X+mar [19].

.")
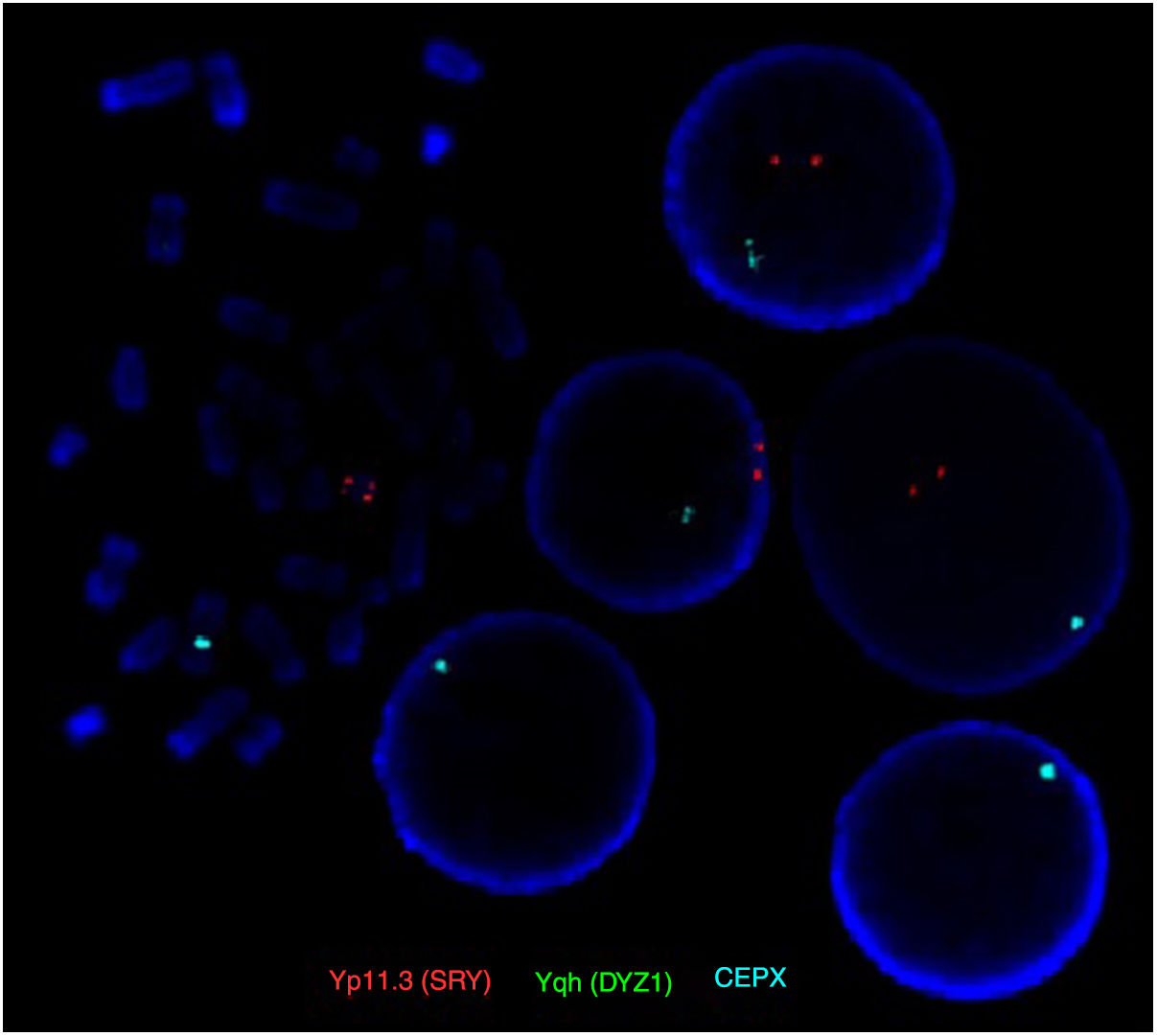
Ante estos hallazgos se amplió la técnica FISH, ya que el contenido de los cromosomas marcadores varía según su procedencia y esto determina su significado clínico. Se hibridaron portas con la sonda CEP que es centromérica para el cromosoma X y para el cromosoma Y, en concreto en la región satélite del centrómero Yp11.1-q11.1 (DYZ3). Se analizaron más de 100 núcleos, observándose una señal de hibridación para el cromosoma X en todos los núcleos, y para la sonda DYZ3 se observa en el 39% de los núcleos dos señales de hibridación, determinando el origen del cromosoma marcador en el cromosoma Y. En la hibridación de la sonda DYZ1 no se obtuvo ninguna señal de hibridación, indicando que no hay presencia de la región heterocromática del cromosoma Y (fig. 3).
 donde se observa una señal de hibridación para la sonda centromérica del cromosoma X (CEPX) color azul, dos señales de hibridación correspondientes al gen SRY (Yp11.3) color rojo. (El color de esta figura solo puede apreciarse en la versión electrónica del artículo).")
Imagen FISH (hibridación in situ fluorecescente) donde se observa una señal de hibridación para la sonda centromérica del cromosoma X (CEPX) color azul, dos señales de hibridación correspondientes al gen SRY (Yp11.3) color rojo. (El color de esta figura solo puede apreciarse en la versión electrónica del artículo).
El gen SRY (Sex-determining region of the Y Chromosome) es el principal determinante testicular, aunque existen evidencias de otros genes implicados en la determinación del sexo5. Gracias a su acción las células germinales de las gónadas del embrión se diferencian y forman tejido testicular, y en ausencia o alteración (mutación, deleción) de este gen, se desarrolla un fenotipo femenino, aunque no siempre se produce este desarrollo sexual ya que son necesarios los dos cromosomas X para que se desarrollen adecuadamente los ovarios y se produzca una adecuada secreción de hormonas sexuales, lo que hace que muchos de estos casos no lleguen a la pubertad sin ayuda hormonal. El gen SRY se encuentra localizado en el brazo corto del cromosoma Y, concretamente en la región Yp11.3. Para la sonda Yp11.3 (SRY) se obtiene una doble señal de hibridación.
Los resultados obtenidos por FISH permitieron definir el cromosoma marcador como un isocromosoma del brazo corto del cromosoma Y, con doble presencia del gen SRY, determinando su fórmula cromosómica final como mos 45,XO/46,Xi(Yp)(SRY++).
Tras presentar el informe citogenético final a Ginecología, se le solicitó una RMN pélvica para el estudio de las gónadas. Aunque en la RMN no se visualizaron anexos ni masas que sugerían presencia de tumor anexial, la paciente fue sometida a una intervención laparoscópica exploradora.
DiscusiónAlgunos casos de DGM quedan sin diagnóstico etiológico definido, ya sea por tener un estudio citogenético incompleto, por la falta de estudio molecular o por estar a la espera de la descripción de un nuevo gen participante en el complejo proceso de desarrollo gonadal. El gen SRY localizado en el cromosoma Y inicia la compleja cascada genética hacia la diferenciación testicular. Los genes implicados que participan tanto en la diferenciación tisular de los diferentes órganos como en la adecuada producción y funcionamiento de las enzimas de la esteroidogénesis no son solo los ubicados en los cromosomas sexuales (SRY, DAX1), sino también genes ubicados en los autosomas (SF1,WT1,SOX9)5,6.
Se han descrito unos 32-40 genes con mutaciones inactivadoras en su mayor parte, aunque también existen anomalías por haploinsuficiencia o por exceso de dosis en alguno de ellos7.
En pacientes con ST con presencia de líneas celulares con cromosoma marcador siempre debe investigarse si existe presencia de secuencias específicas del cromosoma Y; esto adquiere especial relevancia debido al importante riesgo que presentan estos pacientes de desarrollar tumores gonadales8. Los últimos estudios publicados4 sugieren realizar estudios de PCR en linfocitos para poder detectar presencia total o parcial del cromosoma Y sin que sea visible en el estudio citogenético.
El caso que presentamos presentaba el cromosoma Y estructuralmente anormal, cromosoma Y dicéntrico, presente como parte de un mosaico. Parece haber una región entre Yq11 y Yq12 propensa a la rotura; la ruptura de la cromátida hermana y la fusión inadecuada de los extremos rotos podría llevar a formar cromosomas isodicéntricos. Debemos considerar también que en los casos de mosaicismo el porcentaje del mosaico puede variar entre los diferentes tejidos y parece no haber correlación entre los porcentajes de las dos líneas celulares presentes para desarrollar uno u otro fenotipo en estudios de linfocitos. En estudios de tejidos gonadales sí se han correlacionado, viéndose dependencia entre el tipo de gónada y el porcentaje de mosaicismo encontrado en ella.
El riesgo de desarrollo de tumores gonadales aumenta con la edad, por lo que esta paciente presentaba un riesgo considerable debido al retraso de su diagnóstico como DGM.
ConclusionesLos casos de DGM son poco frecuentes y en ellos adquiere especial importancia realizar un adecuado diagnóstico citogenético temprano dado el elevado riesgo de desarrollo de tumores gonadales.
