







La hipercolesterolemia familiar es una enfermedad autosómica dominante que cursa con niveles plasmáticos elevados de colesterol unido a lipoproteínas de baja densidad. La presencia de esta alteración en el metabolismo lipídico se asocia a un aumento del riesgo cardiovascular en los pacientes que la padecen, siendo de gran importancia la realización de un diagnóstico genético para ofrecer un tratamiento adecuado y disminuir la morbimortalidad. El objetivo de este trabajo es describir la aplicación de la secuenciación de nueva generación al diagnóstico genético de la hipercolesterolemia familiar, en comparación con la secuenciación Sanger, la técnica convencional usada hasta el momento.
Material y métodosSe analizaron 110 muestras de sangre venosa periférica procedente de pacientes que presentaban cuadro clínico de hipercolesterolemia familiar mediante secuenciación de nueva generación, utilizando un panel comercial que permite la identificación de mutaciones en los genes LDLR, APOB, PCSK9 y LDLRAP1 (SEQPRO Lipo, Progenika) con la tecnología GS JUNIOR 454 (Roche).
ResultadosAplicando esta tecnología fue posible secuenciar los genes asociados a la hipercolesterolemia familiar descritos hasta el momento en grupos de hasta 20 pacientes simultáneamente. Se detectaron un total de 35 mutaciones en las 110 muestras analizadas, localizándose el 94,29% en el gen LDLR. Todas las mutaciones identificadas fueron confirmadas mediante el método de secuenciación Sanger.
ConclusionesLa utilización de la secuenciación masiva de nueva generación permite la realización de un diagnóstico genético más rápido y un análisis molecular más eficiente de los genes implicados en la hipercolesterolemia familiar con una fiabilidad similar a la técnica convencional Sanger.
Familial hypercholesterolemia is an autosomal dominant disorder that causes increased levels of cholesterol associated with low density lipoproteins in plasma. The presence of altered lipid metabolism in these patients increases their level of risk of suffering from a cardiovascular disease. The certainty of having this genetic disorder by making a timely and precise molecular diagnostic is crucial for the appropriate treatment and the reduction of the disease morbidity-mortality. The aim of this work is to describe the applicability of next generation sequencing technology to the genetic diagnosis of familial hypercholesterolemia and compare this novel method with the conventional Sanger sequencing method.
Material and methodsA next generation sequencing commercial panel (SEQPRO Lipo, Progenika) was used to analyze 110 peripheral venous blood samples from patients with familial hypercholesterolemia. This enables the assessment of mutations in genes associated with the disease (e.g. LDLR, APOB, PCSK9 and LDLRAP1) with the GS JUNIOR 454 technology (Roche).
ResultsApplication of next generation sequencing enables the sequencing of the genes involved in the familial hypercholesterolemias, described so far, in groups of 20 patients simultaneously. Using this novel technology, a total of 35 mutations were detected in the 110 analysed samples, with 94.29% being located in the LDLR gene. Mutations were confirmed by Sanger sequencing.
ConclusionNext generation sequencing enables a quick genetic diagnosis and a more efficient molecular analysis of all genes described so far to be involved in familial hypercholesterolemia, with similar reliability to that of conventional Sanger sequencing.
La hipercolesterolemia familiar (HF; OMIM#143890) es un trastorno genético de herencia autosómica dominante del metabolismo de las lipoproteínas con una prevalencia en la población general alrededor de 1:500 para heterocigotos y 1:1.000.000 para homocigotos1. Este trastorno está caracterizado por un incremento en la concentración plasmática del colesterol unido a lipoproteínas de baja densidad (cLDL), cursando a menudo con el desarrollo de xantomas, aterosclerosis, además de un elevado riesgo de presentar enfermedades coronarias prematuras2. Se han descrito más de 1.500 mutaciones responsables de HF en los genes LDLR, APOB, PCSK9, y LDLRAP1, la mayoría de ellas en el gen LDLR3–7.
El diagnóstico de la HF está basado en una combinación de criterios clínicos, bioquímicos y genéticos8 que han permitido establecer diferentes índices diagnósticos, siendo los criterios de la red de clínicas de lípidos holandesa los más utilizados en España9 (tabla 1). Este índice permite otorgar una estratificación a los pacientes con HF en diagnóstico de certeza (≥8 puntos), probable (6-7 puntos) y posible (3-5 puntos).
Criterios diagnósticos para la hipercolesterolemia familiar basados en la red de clínicas de lípidos holandesas
| Historia familiar | |
| Familiar de primer grado con enfermedad coronaria prematura (hombres<55 años y mujeres<60 años)y/o | 1 |
| Familiar de primer grado con niveles de cLDL>210mg/dlFamiliar de primer grado con xantomas tendinosos y/o arco corneal<45 añosy/oFamiliar<18 años con cLDL ≥ 150mg/dl | 2 |
| Antecedentes personales | |
| Paciente con enfermedad coronaria prematura(hombres<55 años y mujeres<60 años) | 2 |
| Paciente con enfermedad cerebrovascular o arterial periférica prematura (hombres<55 años y mujeres<60 años) | 1 |
| Examen físico | |
| Xantomas tendinososArco corneal<45 años | 64 |
| Análisis de laboratorio | |
| cLDL ≥330mg/dlcLDL 250-329mg/dlcLDL 190-249mg/dlcLDL 155-189mg/dl | 8531 |
| Análisis genético | |
| Mutación funcional en el gen del LDLR, APOB o PCSK9 | 8 |
| Diagnóstico de HF: | |
| Certeza: ≥ 8 puntos; probable: 6-7 puntos; posible: 3-5 puntos | |
La técnica más utilizada hasta el momento para la búsqueda de mutaciones en el ADN es la secuenciación según el método Sanger10. Aunque esta técnica presenta una alta sensibilidad y especificidad, su rendimiento es insuficiente para el cribado de la HF donde es preciso realizar un número muy elevado de secuencias para establecer un diagnóstico genético. Con el desarrollo de la secuenciación de nueva generación («next generation sequencing» [NGS]) el rendimiento de la secuenciación ha aumentado de manera significativa ya que permite secuenciar de forma paralela millones de fragmentos y a un coste mucho menor11,12. La NGS permite la detección de mutaciones puntuales, pequeñas deleciones e inserciones y variaciones en el número de copias (CNV)13. Estas características apuntan a la NGS como una herramienta eficaz para la realización del diagnóstico genético de la HF de forma rápida y económica sin renunciar a una alta especificidad, sensibilidad y precisión14,15.
El objetivo de este trabajo es describir la aplicación de la NGS para la búsqueda de mutaciones en pacientes con HF con el fin de evaluar la sensibilidad de la técnica, a la vez que identificar las ventajas e inconvenientes que esta pueda presentar en el flujo de trabajo en el laboratorio, en comparación con la técnica convencional de secuenciación Sanger.
Material y métodosPacientes incluidos en el estudioSe incluyeron 110 pacientes que presentaban valores de alta probabilidad o certeza diagnóstica clínica de HF basados en los criterios de la red de clínicas de lípidos holandesa, considerándose estos como casos índices. Estas muestras fueron recogidas en el Hospital Clínic de Barcelona durante los años 2012, 2013 y 2014 y la secuenciación de las mismas se realizó en el Servicio de Bioquímica y Genética Molecular del Centro de Diagnóstico Biomédico de este hospital.
Extracción de ADNLa extracción de ADN se realizó a partir de 1ml de sangre venosa de forma automatizada con el extractor QIAsymphony (QIAGEN) utilizando el kit QIAsymphony DSP ADN Kit (Ref. 931255) según el protocolo de la casa comercial (www.qiagen.com). La concentración y pureza de la muestra de ADN fue evaluada mediante espectrofotometría con el Nanodrop 1000 (Thermo Scientific). La concentración óptima de ADN requerida para la NGS se consideró 20ng/μL, con una pureza de entre 1,60-1,95 para OD260/OD280 y alrededor de 1,5 para OD260/OD230.
Metodología «next generation sequencing»Se utilizaron 20μL de ADN de cada muestra a una concentración de 20ng/μL para la amplificación mediante PCR (reacción en cadena de la polimerasa) de las diferentes regiones de los genes: LDLR, APOB, PCSK9 y LDLRAP1. Para ello se utilizaron los cebadores específicos contenidos en el panel SEQPRO Lipo de Progenika (www.progenika.com), siguiendo las indicaciones del fabricante. Las condiciones de amplificación fueron de 94°C por 15 minutos; 23 ciclos de: 94°C por 30 segundos, 65°C por un minuto y 72°C por un minuto y una extensión final de 72°C por 7 minutos, utilizando el termociclador GeneAmp PCR system 9700 de Applied Biosystems. A partir del producto de la PCR de amplificación (amplicones) se realizó una segunda PCR donde se añadieron las etiquetas identificativas (Multiplex Identifiers, MIDs) que permiten la identificación de cada muestra según el paciente de origen. Las condiciones de amplificación de esta segunda PCR fueron: 94°C por 15 minutos; 17 ciclos de: 94°C por 30 segundos, 61°C por un minuto y 72°C por un minuto y una extensión final de 72°C por 10 minutos, utilizando el termociclador descrito anteriormente. La correcta amplificación de las muestras se comprobó mediante visualización por electroforesis en gel de agarosa al 2% de los productos de PCR.
Los amplicones fueron posteriormente purificados mediante el kit Agencourt AMPure XP (Ref. A63880), el cual permite la unión de los amplicones a bolas magnéticas y así eliminar el exceso de adaptadores y cebadores (Protocolo: 000387v001). Una vez purificadas las muestras fueron cuantificadas por fluorimetría usando el kit Quant-iT PicoGreen (Ref. P11496) de ADN de doble cadena, en el equipo Synergy HT (BioTek). La concentración de las muestras en este paso se consideró apta por encima de 10ng/μL. En base a las concentraciones obtenidas, las muestras fueron diluidas hasta una concentración de 109 moléculas/μL, a partir de la cual se realizó una mezcla de todas las muestras de la carrera en un mismo tubo en una cantidad equimolar (pool de amplicones). El pool de amplicones se diluyó finalmente hasta una concentración de 106 moléculas/μL. A continuación, se procedió a la clonación mediante PCR en emulsión (emPCR). El objetivo de la emPCR es realizar una amplificación clonal de la librería para que la señal sea detectable en la secuenciación16,17. Para conseguir esto, se realiza una emulsión de agua en aceite en la que se obtienen millones de microgotas aisladas por una fase lipídica, cada una de las cuales contiene los reactivos de la PCR y una bola de captura («bead») unida a un único amplicón de cadena sencilla. Cada micela de la emulsión funciona como un microrreactor, dentro del cual tiene lugar la amplificación clonal. Las condiciones de amplificación para la emPCR fueron de 94°C por 4 minutos; 50 ciclos de: 94°C por 30 segundos, 58°C por 4,5 minutos y 68°C por 30 segundos, utilizando el termociclador GeneAmp PCR system 9700. Una vez finalizada la emPCR el producto final fue recogido en un falcón de 50mL donde se llevó a cabo la rotura de la emulsión, el lavado, la recuperación y el enriquecimiento de las beads cargadas de ADN. El resultado del enriquecimiento fue evaluado mediante GS Bead Counter y se confirmó un enriquecimiento de beads de entre 0,5×106 y 2×106 en todas las carreras que se realizaron. A continuación sobre un total de 0,5×106 beads se realizó la ligación de los primers A y B para la secuenciación y se añadieron las beads con el ADN control. Todo ello fue realizado con el kit GS Junior Titanium emPCR Kit Lib-A (Ref. 05996520001) siguiendo las instrucciones de la casa comercial Roche (Protocolo: emPCR Amplification Manual Lib-A).
Finalmente, la secuenciación fue realizada utilizando los kits GS Junior Titanium Sequencing Kit (Ref. 05996554001) y GS Junior Titanium PicoTiter Plate Kit (Ref. 05996619001) en el ultrasecuenciador GS Junior 454 siguiendo las indicaciones de la casa comercial Roche (Protocolo: Sequencing Method Manual). Se utilizó el programa de secuenciación de 200 ciclos para la obtención de secuencias de 400 bases aproximadamente. El ultrasecuenciador GS Junior 454 tiene como principio la pirosecuenciación, técnica basada en la liberación de los pirofosfatos que se producen durante la reacción de síntesis del ADN. Mediante una serie de reacciones enzimáticas los pirofosfatos son transformados en un haz de luz que es detectado por una cámara CCD y las señales son procesadas por un sistema de análisis de datos que permite la identificación de todos los nucleótidos incorporados a la cadena en cada ciclo de la secuenciación, permitiendo así conocer la secuencia de estudio16–19.
Una vez finalizada cada carrera de secuenciación se revisó el control de calidad de las secuencias siguiendo los parámetros establecidos por la casa comercial Progenika y se prepararon las muestras para el análisis bioinformático por parte del especialista.
Metodología SangerCon el fin de determinar la sensibilidad y fiabilidad de la NGS, todas las mutaciones encontradas fueron confirmadas mediante la técnica convencional de secuenciación Sanger. La amplificación del fragmento de interés fue realizada mediante una PCR clásica, usando dNTPs (Ref. 11969064001) y Taq Polimerasa (Ref. 11146165001) ambos de Roche y los cebadores específicos de la región a amplificar con condiciones de amplificación específicas para cada región. Estas reacciones se llevaron a cabo en el termociclador GeneAmp PCR system 9700. El producto obtenido fue purificado para eliminar los cebadores y nucleótidos no incorporados, usando las EXCELAPURE 96-Well UF PCR Purification plates de EdgeBio (Ref: 36181). El producto purificado fue sometido a electroforesis en gel de agarosa al 2% para confirmar que el tamaño del fragmento amplificado era el correcto.
A partir del producto purificado se realizó la PCR de secuenciación usando el kit Big Dye Terminator v.3.1 (Ref. 4337455) de Applied Biosystems, basado en el método Sanger, en el cual se incorporan nucleótidos dideoxido (ddNTPs) marcados con fluorescencia a una nueva cadena de ADN sintetizada por una polimerasa. Estos ddNTPs carecen de uno de los grupos hidroxilo y al producirse dicha incorporación se produce la terminación de la cadena, generando así fragmentos de diferente longitud con el último nucleótido marcado10,20. Una vez finalizada la PCR de secuenciación se realizó la purificación de los productos usando las PERFORMA DTR V3 96-Well Short Plates de EdgeBio (Ref: 63887) para eliminar los ddNTPs no incorporados y evitar interferencias en el análisis.
Finalmente, el producto obtenido fue cargado en un secuenciador automático (Genetic Analyzer 3130XL, Applied Biosystems) para separar los fragmentos mediante electroforesis capilar. Este secuenciador produce una inyección electrocinética para que las moléculas puedan entrar en el capilar y migren por él hasta llegar al láser que se encarga de excitar el fluorocromo. De esta manera, la cámara capta la señal emitida transformándola en un electroferograma que permite conocer la secuencia de la cadena20.
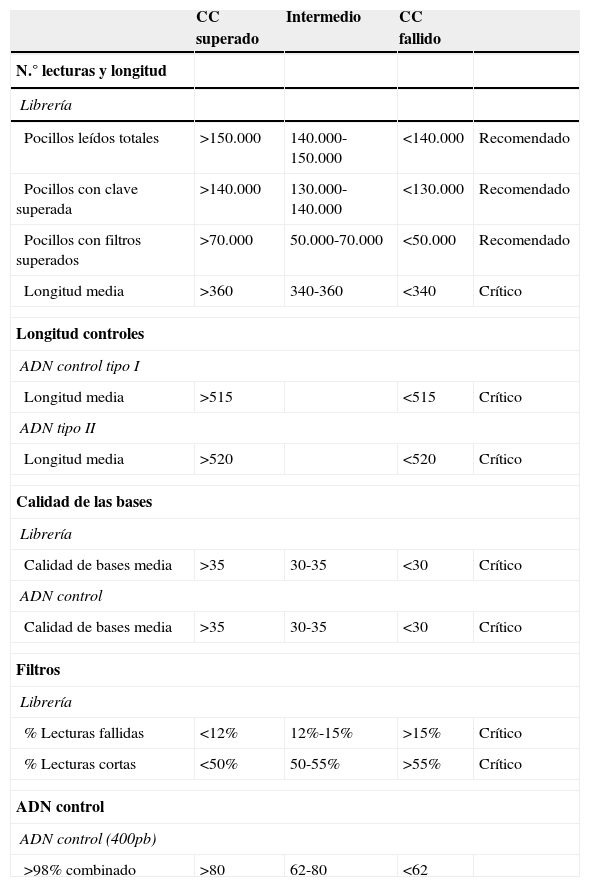
Resultados«Next generation sequencing»Se analizaron mediante NGS 110 muestras de pacientes considerados casos índice de HF, utilizando la tecnología GS Junior 454 de Roche. Las muestras fueron distribuidas en 8 carreras de secuenciación obteniéndose un promedio de 82.200 lecturas/carrera, el rendimiento de las cuales fue de hasta 40 Mb con una cobertura por región de 80-100x, siendo la cobertura mínima aceptada en las regiones con variantes de 15x. Los controles de calidad usados para validar cada carrera fueron los siguientes; la obtención de un mínimo de 70.000 lecturas (Passed Filter Wells) con una media de 360 bases de longitud (Length Average), (fig. 1), una longitud de los ADN control superior a 515 (control tipo I) y 520 bases (control tipo II), (fig. 2A y B) y un porcentaje de lecturas fallidas (Failed: % Dot±Mixed) y de corta longitud (Failed: % Short) no superior al 12 y al 50%, respectivamente (fig. 3). Todos estos parámetros fueron superados en todas las carreras realizadas. En la tabla 2 se muestran los diferentes apartados de este control de calidad.
 de la librería de amplicones obtenida con el kit SEQPRO Lipo, en una carrera de NGS realizada con el ultrasecuenciador GS 454 de Roche.")
 Resultados del control de calidad del ADN Control tipo I, en una carrera de NGS realizada con el ultrasecuenciador GS 454 de Roche. B) Resultados del control de calidad del ADN Control tipo Il, en una carrera de NGS realizada con el ultrasecuenciador GS 454 de Roche.")
 de la librería de amplicones obtenida con el kit SEQPRO Lipo, en una carrera de NGS realizada con el ultrasecuenciador GS 454 de Roche.")
Parámetros de control de calidad de la carrera de secuenciación establecidos por Progenika
| CC superado | Intermedio | CC fallido | ||
|---|---|---|---|---|
| N.° lecturas y longitud | ||||
| Librería | ||||
| Pocillos leídos totales | >150.000 | 140.000-150.000 | <140.000 | Recomendado |
| Pocillos con clave superada | >140.000 | 130.000-140.000 | <130.000 | Recomendado |
| Pocillos con filtros superados | >70.000 | 50.000-70.000 | <50.000 | Recomendado |
| Longitud media | >360 | 340-360 | <340 | Crítico |
| Longitud controles | ||||
| ADN control tipo I | ||||
| Longitud media | >515 | <515 | Crítico | |
| ADN tipo II | ||||
| Longitud media | >520 | <520 | Crítico | |
| Calidad de las bases | ||||
| Librería | ||||
| Calidad de bases media | >35 | 30-35 | <30 | Crítico |
| ADN control | ||||
| Calidad de bases media | >35 | 30-35 | <30 | Crítico |
| Filtros | ||||
| Librería | ||||
| % Lecturas fallidas | <12% | 12%-15% | >15% | Crítico |
| % Lecturas cortas | <50% | 50-55% | >55% | Crítico |
| ADN control | ||||
| ADN control (400pb) | ||||
| >98% combinado | >80 | 62-80 | <62 | |
CC: control de calidad.
El análisis de los resultados fue realizado con el software LipoNext LNX Analysis Software, el cual transforma los datos brutos del sistema Junior en genotipos y analiza las secuencias proporcionando las variantes encontradas y su estado de patogenicidad, siempre que este sea conocido. Todos los resultados fueron validados por el especialista.
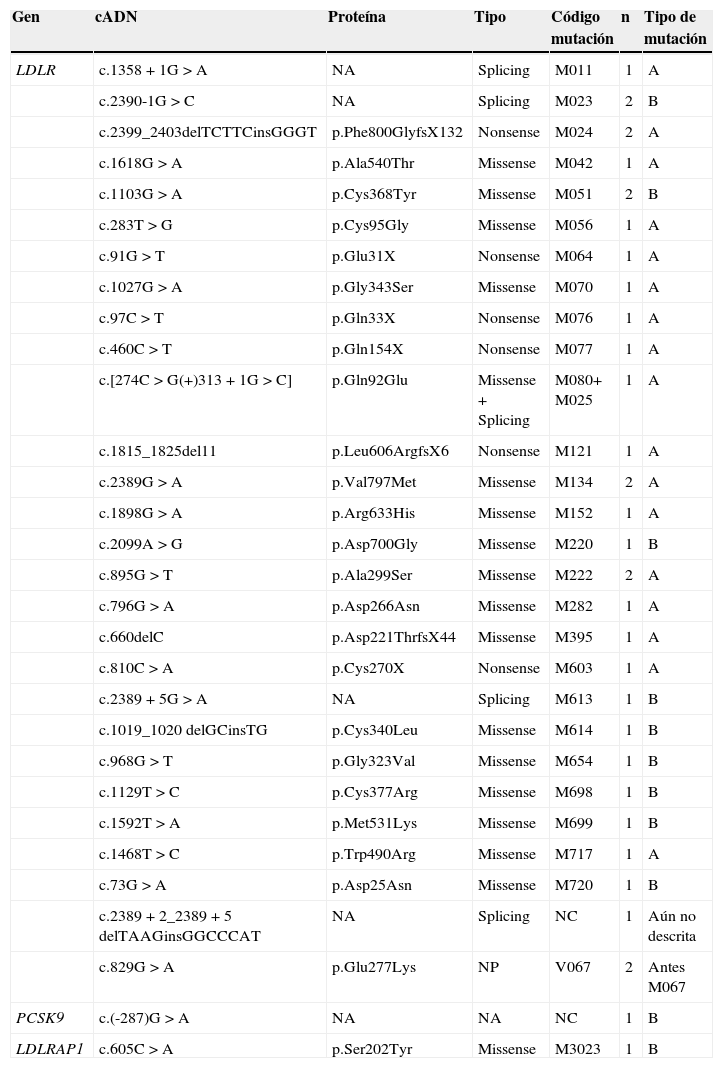
Mutaciones detectadasUn total de 33 de las 110 muestras analizadas por NGS presentaron variantes clasificadas como patogénicas (mutaciones asociadas al riesgo de desarrollar HF). El 93,94% (31/33) de dichas muestras presentaron una única variante en heterocigosis, uno de los pacientes presentó una mutación en el gen LDLR y otra en el gen PCSK9, mientras que otro resultó ser un heterocigoto compuesto presentando dos mutaciones en el gen LDLR. En total fueron identificadas 35 mutaciones en 33 pacientes.
El 94,29% (33/35) de estas mutaciones se encontraron en el gen LDLR, de las cuales el 60,61% (20/33) correspondían a un cambio de sentido o sentido erróneo (missense), el 21,21% (7/33) correspondían a la formación de un codón de terminación (nonsense) y el 18,18% (6/33) afectaban la maduración (splicing) del ARN. Las dos mutaciones restantes se hallaron en los genes PCSK9 (localizada en una región no codificante) y LDLRAP1 (mutación tipo missense). Las mutaciones encontradas y su clasificación se muestran en la tabla 3.
Variantes encontradas en el estudio, previamente identificadas como patogénicas y su clasificación mutacional
| Gen | cADN | Proteína | Tipo | Código mutación | n | Tipo de mutación |
|---|---|---|---|---|---|---|
| LDLR | c.1358+1G>A | NA | Splicing | M011 | 1 | A |
| c.2390-1G>C | NA | Splicing | M023 | 2 | B | |
| c.2399_2403delTCTTCinsGGGT | p.Phe800GlyfsX132 | Nonsense | M024 | 2 | A | |
| c.1618G>A | p.Ala540Thr | Missense | M042 | 1 | A | |
| c.1103G>A | p.Cys368Tyr | Missense | M051 | 2 | B | |
| c.283T>G | p.Cys95Gly | Missense | M056 | 1 | A | |
| c.91G>T | p.Glu31X | Nonsense | M064 | 1 | A | |
| c.1027G>A | p.Gly343Ser | Missense | M070 | 1 | A | |
| c.97C>T | p.Gln33X | Nonsense | M076 | 1 | A | |
| c.460C>T | p.Gln154X | Nonsense | M077 | 1 | A | |
| c.[274C>G(+)313+1G>C] | p.Gln92Glu | Missense+Splicing | M080+ M025 | 1 | A | |
| c.1815_1825del11 | p.Leu606ArgfsX6 | Nonsense | M121 | 1 | A | |
| c.2389G>A | p.Val797Met | Missense | M134 | 2 | A | |
| c.1898G>A | p.Arg633His | Missense | M152 | 1 | A | |
| c.2099A>G | p.Asp700Gly | Missense | M220 | 1 | B | |
| c.895G>T | p.Ala299Ser | Missense | M222 | 2 | A | |
| c.796G>A | p.Asp266Asn | Missense | M282 | 1 | A | |
| c.660delC | p.Asp221ThrfsX44 | Missense | M395 | 1 | A | |
| c.810C>A | p.Cys270X | Nonsense | M603 | 1 | A | |
| c.2389+5G>A | NA | Splicing | M613 | 1 | B | |
| c.1019_1020 delGCinsTG | p.Cys340Leu | Missense | M614 | 1 | B | |
| c.968G>T | p.Gly323Val | Missense | M654 | 1 | B | |
| c.1129T>C | p.Cys377Arg | Missense | M698 | 1 | B | |
| c.1592T>A | p.Met531Lys | Missense | M699 | 1 | B | |
| c.1468T>C | p.Trp490Arg | Missense | M717 | 1 | A | |
| c.73G>A | p.Asp25Asn | Missense | M720 | 1 | B | |
| c.2389+2_2389+5 delTAAGinsGGCCCAT | NA | Splicing | NC | 1 | Aún no descrita | |
| c.829G>A | p.Glu277Lys | NP | V067 | 2 | Antes M067 | |
| PCSK9 | c.(-287)G>A | NA | NA | NC | 1 | B |
| LDLRAP1 | c.605C>A | p.Ser202Tyr | Missense | M3023 | 1 | B |
A: mutaciones directamente asociadas con la HF ya fuera por previa validación in vitro o porque las mutaciones producían un alelo nulo; B: mutaciones asociadas con la HF en otras poblaciones o asociadas con el fenotipo de la enfermedad en estudios familiares pero sin validación in vitro; n: número de pacientes; NA: no aplica; NC: no codificada; NP: no patogénica.
De las muestras con variantes patogénicas, el 58,82% (20/34) eran portadoras de mutaciones directamente asociadas con la HF ya fuera por previa validación in vitro o porque las mutaciones producían un alelo nulo (clasificación mutacional A). El 38,24% (13/34) de las muestras eran portadoras de mutaciones asociadas con la HF en otras poblaciones o asociadas con el fenotipo de la enfermedad en estudios familiares pero sin validación in vitro (clasificación mutacional B). En una de las muestras se localizó una variante de la cual se desconocía la patogenicidad. Por otra parte, 2 muestras de las 110 analizadas presentaron la variante V067 (c.829G>A p.Glu277Lys) considerada anteriormente como patogénica, aunque actualmente se considera un polimorfismo. Con el fin de simplificar los resultados, las variantes encontradas cuya patogenicidad no se encuentra aún validada no son mencionadas en este trabajo.
Validación del estudioPara validar el estudio todas las mutaciones encontradas por NGS fueron comprobadas por secuenciación Sanger confirmándose en el 100% de los casos. En la figura 4 se muestra un ejemplo de una mutación detectada mediante ambas técnicas.
, detectada por ambas técnicas. Sanger, panel superior, mostrando electroferograma y NGS, panel inferior, con la tabla obtenida mediante el software LipoNext LNX Analysis. También se puede observar la variante no patogénica c.81C>T (rosa). El color de esta figura solo puede apreciarse en la versión electrónica del artículo.")
Ejemplo de la mutación M064, c.91G>T (verde), detectada por ambas técnicas. Sanger, panel superior, mostrando electroferograma y NGS, panel inferior, con la tabla obtenida mediante el software LipoNext LNX Analysis. También se puede observar la variante no patogénica c.81C>T (rosa). El color de esta figura solo puede apreciarse en la versión electrónica del artículo.
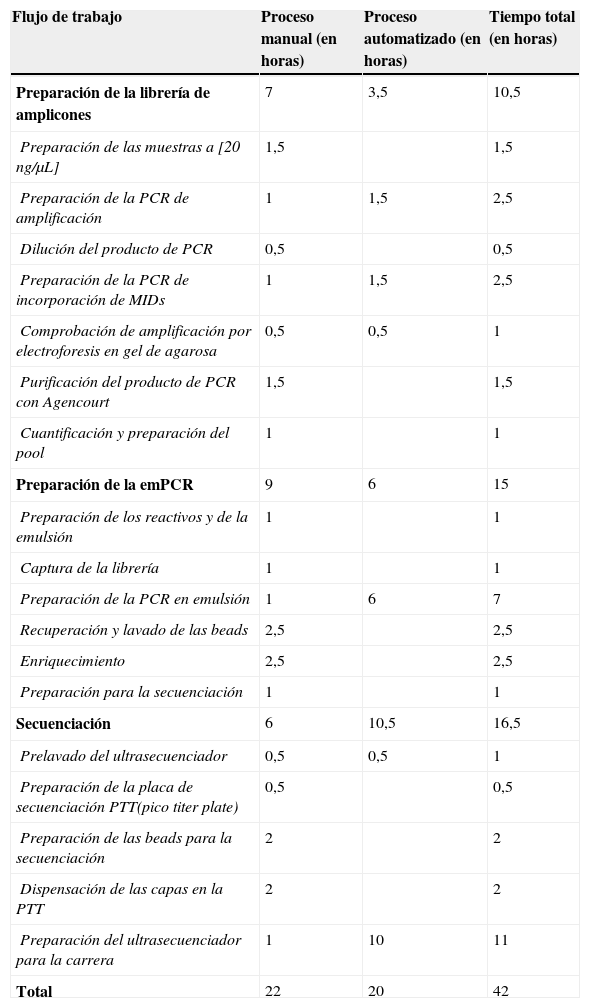
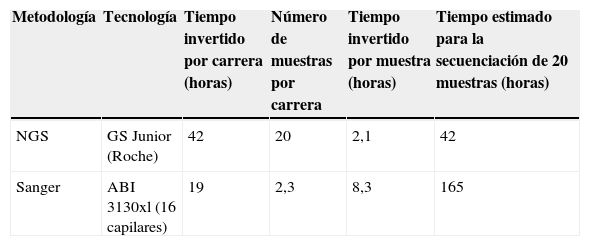
En concreto, en una sola carrera de NGS fue posible analizar los 18 exones del gen LDLR, 2 de los 26 exones del gen APOB, los 12 exones del gen PCSK9 y los 9 exones del gen LDLRAP1. En total 41 exones secuenciados de manera bidireccional por paciente y hasta 20 pacientes por carrera, lo que da un total de 820 exones secuenciados en tan solo 42 horas de trabajo de laboratorio. Esto significa que para secuenciar el mismo número de exones por la técnica de Sanger se necesitarían 165 horas de trabajo de laboratorio (aproximadamente 20 días).
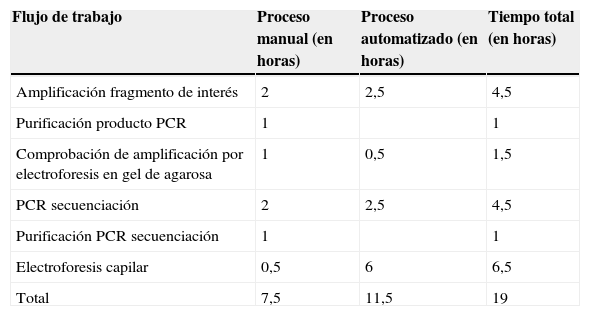
El flujo de trabajo y el tiempo invertido en cada paso del proceso se muestra en la tabla 4 (NGS) y tabla 5 (secuenciación Sanger), así como el tiempo estimado para la secuenciación de los genes implicados en la HF por ambas técnicas (tabla 6).
Flujo de trabajo de la NGS y tiempo en horas invertido en promedio en cada paso del proceso de una carrera de secuenciación utilizando el ultrasecuenciador GS Junior 454 de Roche
| Flujo de trabajo | Proceso manual (en horas) | Proceso automatizado (en horas) | Tiempo total (en horas) |
|---|---|---|---|
| Preparación de la librería de amplicones | 7 | 3,5 | 10,5 |
| Preparación de las muestras a [20ng/μL] | 1,5 | 1,5 | |
| Preparación de la PCR de amplificación | 1 | 1,5 | 2,5 |
| Dilución del producto de PCR | 0,5 | 0,5 | |
| Preparación de la PCR de incorporación deMIDs | 1 | 1,5 | 2,5 |
| Comprobación de amplificación por electroforesis en gel de agarosa | 0,5 | 0,5 | 1 |
| Purificación del producto de PCR con Agencourt | 1,5 | 1,5 | |
| Cuantificación y preparación del pool | 1 | 1 | |
| Preparación de la emPCR | 9 | 6 | 15 |
| Preparación de los reactivos y de la emulsión | 1 | 1 | |
| Captura de la librería | 1 | 1 | |
| Preparación de la PCR en emulsión | 1 | 6 | 7 |
| Recuperación y lavado de las beads | 2,5 | 2,5 | |
| Enriquecimiento | 2,5 | 2,5 | |
| Preparación para la secuenciación | 1 | 1 | |
| Secuenciación | 6 | 10,5 | 16,5 |
| Prelavado del ultrasecuenciador | 0,5 | 0,5 | 1 |
| Preparación de la placa de secuenciaciónPTT(pico titer plate) | 0,5 | 0,5 | |
| Preparación de las beads para la secuenciación | 2 | 2 | |
| Dispensación de las capas en la PTT | 2 | 2 | |
| Preparación del ultrasecuenciador para la carrera | 1 | 10 | 11 |
| Total | 22 | 20 | 42 |
MIDs: identificadores de la multiplex; PCR: reacción en cadena de la polimerasa; PTT: pico titer plate.
Flujo de trabajo de la secuenciación Sanger y tiempo en horas invertido en promedio en cada paso del proceso utilizando el analizador automático ABI 3130xl de Applied Biosystems (16 capilares, placa de 96)
| Flujo de trabajo | Proceso manual (en horas) | Proceso automatizado (en horas) | Tiempo total (en horas) |
|---|---|---|---|
| Amplificación fragmento de interés | 2 | 2,5 | 4,5 |
| Purificación producto PCR | 1 | 1 | |
| Comprobación de amplificación por electroforesis en gel de agarosa | 1 | 0,5 | 1,5 |
| PCR secuenciación | 2 | 2,5 | 4,5 |
| Purificación PCR secuenciación | 1 | 1 | |
| Electroforesis capilar | 0,5 | 6 | 6,5 |
| Total | 7,5 | 11,5 | 19 |
PCR: reacción en cadena de la polimerasa.
Tiempo estimado según la metodología aplicada, para la secuenciación de los genes implicados en la HF (gen LDLR: 18 exones, gen APOB 2/26 exones, gen PCSK9: 12 exones y gen LDLRAP1: 9 exones, en total 41 exones por muestra)
| Metodología | Tecnología | Tiempo invertido por carrera (horas) | Número de muestras por carrera | Tiempo invertido por muestra (horas) | Tiempo estimado para la secuenciación de 20 muestras (horas) |
|---|---|---|---|---|---|
| NGS | GS Junior (Roche) | 42 | 20 | 2,1 | 42 |
| Sanger | ABI 3130xl (16 capilares) | 19 | 2,3 | 8,3 | 165 |
La confirmación mediante Sanger del 100% de las mutaciones encontradas con la técnica NGS aquí descrita, demuestra la alta sensibilidad y especificidad de la misma para el diagnóstico genético de la HF. Estos resultados coinciden con un estudio realizado por Maglio et al., en el cual se encontró una sensibilidad del 99,6%, una especificidad del 100% y una precisión del 99,6% en la metodología NGS para la detección de mutaciones puntuales21. En dicho estudio, realizado en la población sueca, se encontró que el 90% de las mutaciones identificadas pertenecían al gen LDLR y el 10% restante se localizaban en los genes APOB y PCSK9 (8% y 2% respectivamente). Estos resultados muestran, al igual que los obtenidos en este estudio, que el gen donde se encuentra la mayor proporción de mutaciones es el LDLR (94,29%), mientras que la proporción es minoritaria en los otros genes implicados. No se observaron coincidencias en el tipo de mutaciones encontradas en ambos estudios, pero la alta variabilidad de las mutaciones encontradas en este estudio corrobora la gran heterogeneidad mutacional del gen LDLR presente en la población española, tal y como describió Palacios et al.22.
En resumen, la principal ventaja de la aplicación de la NGS al diagnóstico de la HF es la posibilidad de secuenciar todos los genes asociados a la enfermedad en una sola carrera de secuenciación, es decir: los 18 exones del gen LDLR, 2 de los 26 exones del gen APOB, los 12 exones del gen PCSK9 y los 9 exones del gen LDLRAP1. La aplicación de la NGS al cribado de la HF resulta en una disminución del tiempo de trabajo de laboratorio y en una reducción de los costes como consecuencia de esta menor carga laboral, de hecho autores como Sharma et al. han postulado a la NGS como la herramienta más eficiente y económica para el diagnóstico genético de la HF23. Este abaratamiento de la secuenciación ha llevado a la NGS a convertirse en la técnica de elección para el diagnóstico genético de un gran número de patologías tales como: la distrofia corneal y retinopatías24,25, el análisis de los genes BRCA1 y BRCA2 en cáncer de mama26,27, la esclerosis múltiple28, así como el análisis de la resistencia a antirretrovirales en VIH29, entre otras.
Por otro lado, una limitación de la aplicación de la NGS para el diagnóstico de la HF en el laboratorio es la necesidad de realizar cada carrera de secuenciación con un número igual o superior a 12 muestras; de no ser así los costes por muestra secuenciada aumentan significativamente y no se consigue el máximo rendimiento de la técnica. Esto puede retrasar el tiempo de realización de la técnica y por consiguiente el tiempo de respuesta si no se dispone de un número suficiente de casos índices en un periodo determinado de tiempo para realizarla.
Otra limitación de la técnica es debida al principio de pirosecuenciación que esta utiliza, ya que puede presentar errores de secuenciación en las regiones ricas en homopolímeros.
Finalmente, es importante mencionar que aunque la NGS es una herramienta eficaz para el diagnóstico de la HF no siempre es posible identificar mutaciones en los pacientes afectos, esto puede ser debido a que existen otros loci asociados a la enfermedad aún sin identificar. Además, el kit SEQPRO Lipo aquí utilizado solo cubre dos exones del gen APOB, mientras que autores como Vandrovcova et al. recomiendan secuenciar el gen APOB por completo para realizar un diagnóstico más exhaustivo de la enfermedad14.
Con los datos obtenidos se concluyó que, aunque la secuenciación tanto por Sanger como por NGS permiten una detección específica de las mutaciones asociadas a la HF, por motivos económicos, de carga laboral y de rapidez en la obtención de los resultados, el método NGS es el más indicado en el estudio de los casos índice. Por otro lado para el estudio de casos familiares, donde se busca una mutación ya conocida, el método Sanger sigue siendo la opción más adecuada. Aplicando este algoritmo se consigue agilizar significativamente el flujo de trabajo en el laboratorio y disminuye el tiempo de respuesta.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Queremos agradecer al Servicio de Bioquímica y Genética Molecular del Hospital Clínic de Barcelona, en especial a Vanesa López Álvarez, Loli Jiménez Sánchez y María Isabel Álvarez-Mora, su colaboración en la elaboración del manuscrito.
