


La irrupción de la proteómica en la última década ha abierto nuevas perspectivas en la investigación clínica, principalmente en la búsqueda de nuevos biomarcadores de diagnóstico, pronóstico, recuperación y respuesta a un tratamiento farmacológico. En su aplicación clínica, la proteómica está llamada a revolucionar la definición actual de enfermedad, al desarrollar un nuevo concepto que englobe el conjunto de cambios fisiológicos y patológicos derivados de una enfermedad y estableciendo perfiles proteicos que permitan realizar un diagnóstico más preciso. En este artículo se recogen, mediante una descripción detallada, las principales aportaciones que la proteómica ofrece a la práctica clínica.
The irruption of proteomics in the last decade has opened new insights in clinical research, mainly in the search for new biomarkers of diagnosis, prognosis and recovery, as well as the response to pharmacological treatments. In its clinical application, proteomics is beginning to revolutionise the current definition of disease, developing a new concept that includes both the physiological changes and pathological origins of a disease, and establishing proteomic profiles that allow the clinician to make a more precise diagnosis. In this article presents a detailed description of the main contributions proteomics offers to clinical practice.
El análisis de proteínas en los diferentes fluidos biológicos constituye una de las aproximaciones más antiguas e importantes llevadas a cabo en los laboratorios clínicos. En la actualidad, las proteínas son junto con metabolitos de diferente naturaleza, los dos grupos de analitos más utilizados en la práctica clínica para el diagnóstico de diversas enfermedades. Además de la cuantificación de proteínas individuales, se ha venido proponiendo la determinación de perfiles proteicos cuyas proteínas puedan ser valoradas conjuntamente para que puedan tener utilidad diagnóstica.
En este sentido, la proteómica constituye un paso adelante respecto al análisis clínico convencional de medida de parámetros individuales y previamente establecidos, ya que su objetivo principal consiste en el análisis global cualitativo y cuantitativo de todas las proteínas que componen un sistema biológico definido en un momento determinado.
¿Qué es un biomarcador?En el año 2001 el National Institutes of Health (NIH) definió el término de biomarcador como «una característica que se mide y evalúa objetivamente como indicador de procesos biológicos normales, procesos patogénicos o respuestas farmacológicas a una intervención terapéutica»1. Desde el punto de vista proteómico, un biomarcador será por tanto una proteína o grupo de proteínas cuya variación de abundancia permita establecer un diagnóstico en una fase temprana de las enfermedades, un pronóstico acerca de su desarrollo y/o una evaluación de la respuesta a un tratamiento. Se sabe con certeza que cualquier enfermedad conlleva la alteración de proteínas concretas, las cuales a su vez pueden provocar modificaciones sobre otras proteínas de modo que se produzca una cascada de alteraciones y donde un elevado número de proteínas se verán implicadas. Por ello, el estudio simultáneo del proteoma en su conjunto, sin sesgo o preselección de moléculas objeto de investigación, será de gran utilidad para una mejora en la medicina del futuro.
La proteómica y su aplicación clínicaEl término proteoma fue utilizado por primera vez por Wilkins et al en 1995 al describir la composición proteica total de un organismo a partir de la determinación de todas las proteínas producidas por su ADN2. Derivado de este concepto nació el término proteómica para denominar a la disciplina encargada del estudio del proteoma3. El mayor desarrollo de la proteómica se inició en el año 2001 con la creación de la Human Proteome Organization (HUPO), cuya finalidad es contribuir al desarrollo de la proteómica mediante la estimulación de la investigación y la difusión de conocimiento del proteoma humano4.
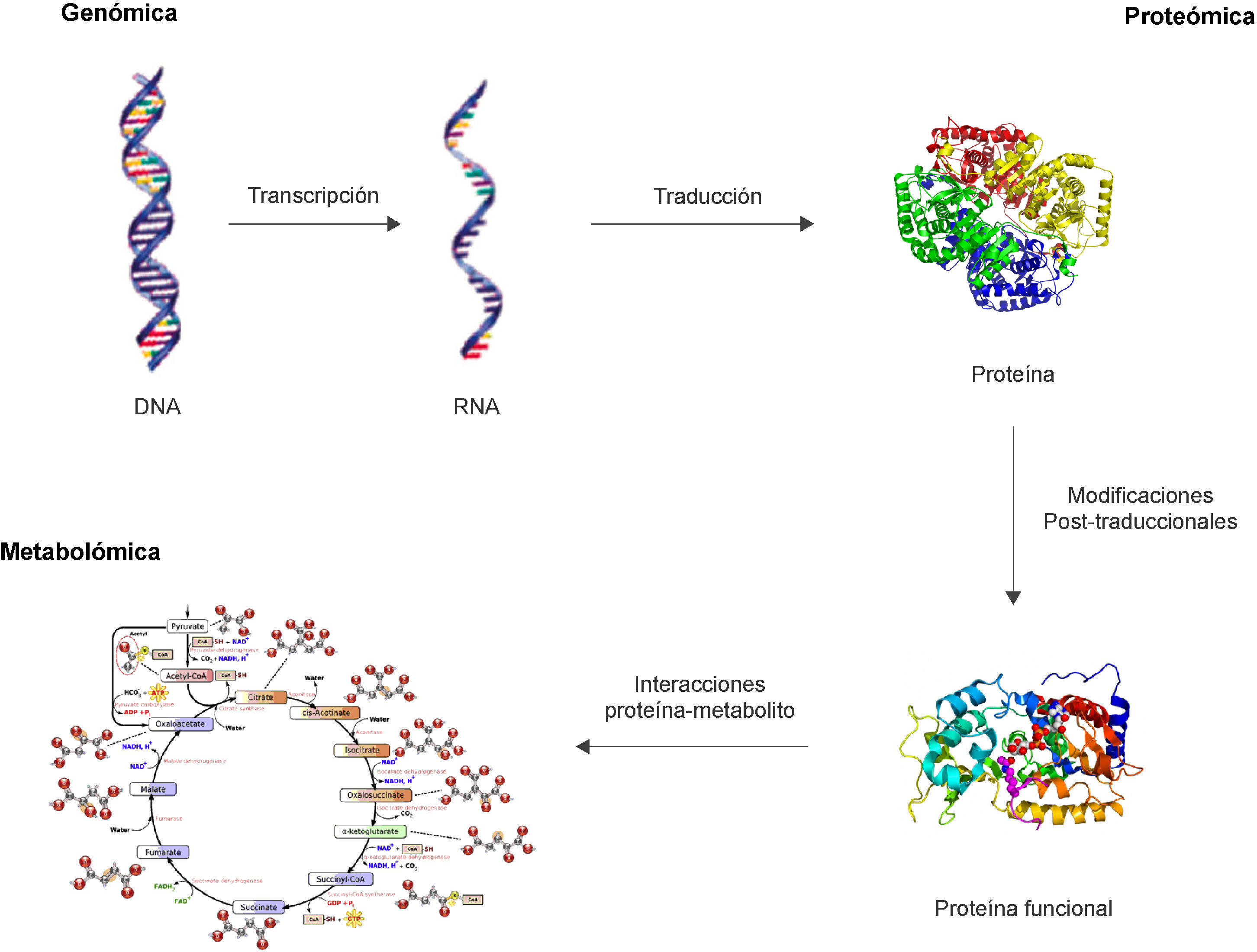
El proteoma, a diferencia del genoma, es un concepto dinámico en el que se incluyen el conjunto de todas las proteínas presentes en un sistema biológico (célula, organismo, etc.) incluyendo tanto las proteínas traducidas directamente a partir del material genético, como también todas aquellas proteínas sometidas a modificaciones pos-transcripcionales y pos-traduccionales5 (fig. 1).

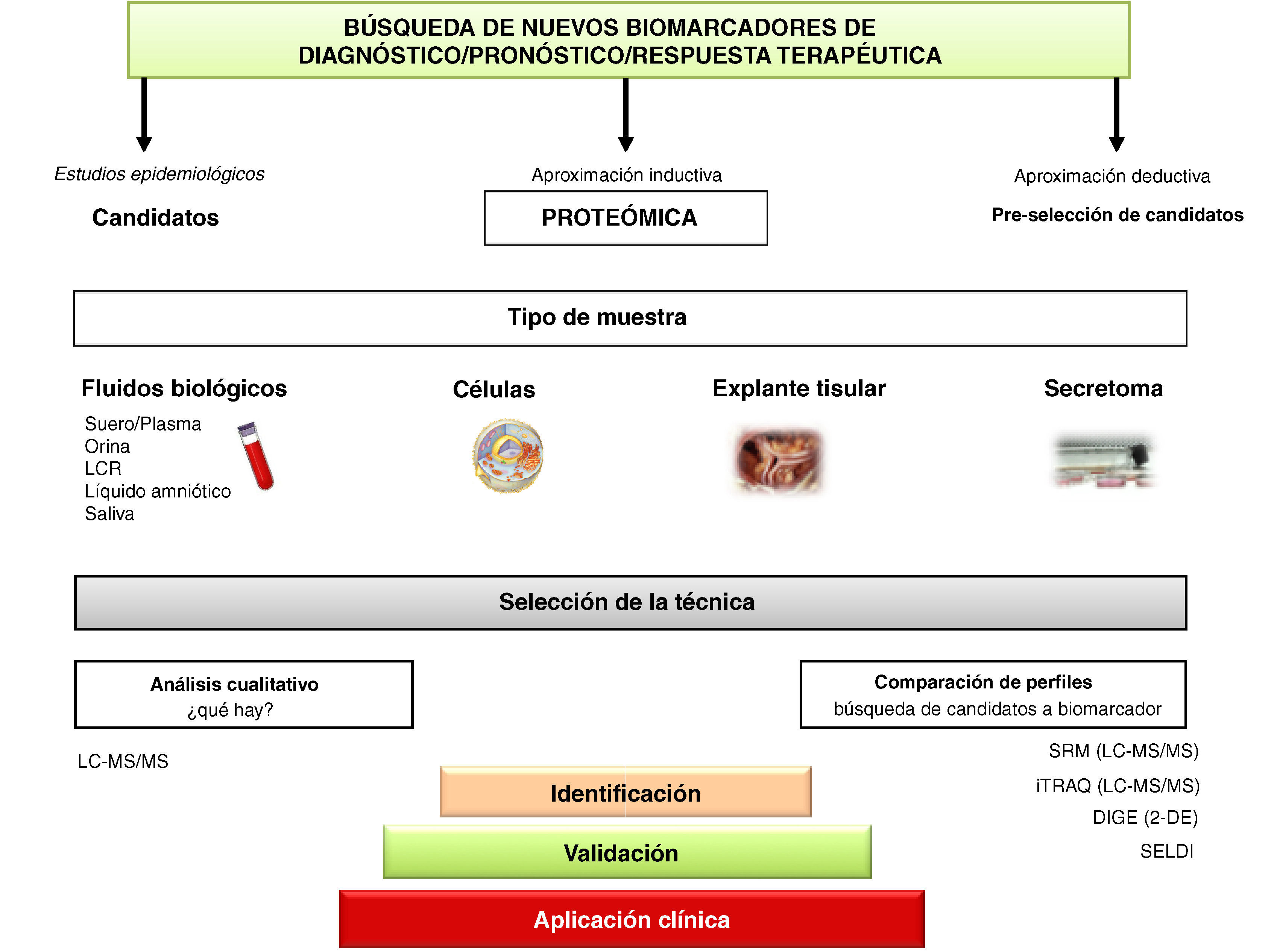
En los estudios de proteómica se puede detectar la composición/estructura de las proteínas, los cambios conformacionales, las alteraciones moduladoras durante el desarrollo y las modificaciones pos-traduccionales. El estudio del proteoma permite, así mismo, conocer variaciones cuantitativas de las proteínas dependiendo de su estado de desarrollo, el gasto metabólico, el estado fisiopatológico del organismo, etc., así como las interacciones con otras proteínas o con fármacos. En este sentido y puesto que las proteínas se encuentran organizadas y expresadas en sistemas que interactúan entre sí, su estudio puede llegar a ser muy complicado debido a las múltiples interacciones que éstas tienen dentro del sistema. La proteómica permite, además, identificar proteínas individuales en mezclas complejas de miles de moléculas a partir de cantidades mínimas de muestra biológica. Dentro de la proteómica existen distintas sub-especialidades: 1) proteómica de abundancia, en la que se realiza un estudio cualitativo y cuantitativo de los niveles proteicos6; 2) proteómica funcional que se centra en el estudio de funciones específicas de las proteínas en diferentes condiciones fisiopatológicas7; 3) proteómica estructural, la cual trata de caracterizar las estructuras proteicas individuales con el fin de comprender su función celular basándose en el análisis tridimensional y la realización de modelos de proteínas8, y 4) proteómica clínica que surge como nueva disciplina basada en la aplicación de las técnicas y estrategias proteómicas al diagnóstico, pronóstico, tratamiento y seguimiento de las enfermedades humanas (fig. 2).
Técnicas proteómicas en medicina traslacional. La búsqueda de biomarcadores en las distintas muestras biológicas disponibles pasa por la selección o combinación de las técnicas más adecuadas hasta llegar a su identificación y validación.")
Existe un amplio abanico de técnicas y metodologías aplicadas en proteómica. En el apartado siguiente se resumen los fundamentos de la 2-DE, por su gran aplicabilidad a las distintas muestras biológicas disponibles, y de varias metodologías de reciente aparición orientadas a la cuantificación sensible de las variaciones en los niveles de abundancia proteica.
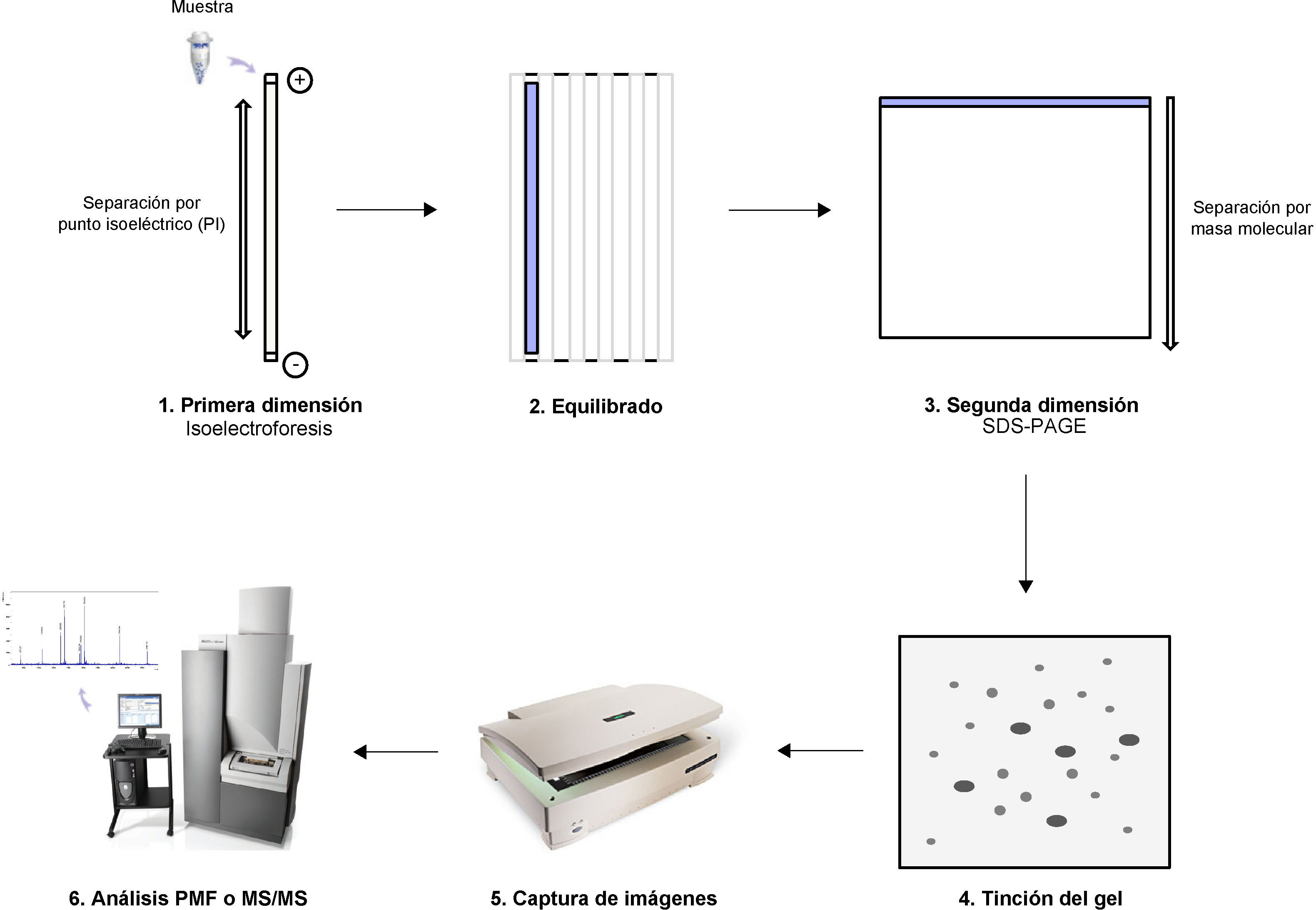
Electroforesis bidimensional convencional en gel (2-DE)La electroforesis bidimensional (2-DE) es una técnica de separación de proteínas que se basa en dos etapas de separación: isoelectroenfoque y SDS-PAGE9,10. En la primera, la mezcla proteica se separa en un gradiente de pH (pH 4-7, pH 3-10, etc.) de acuerdo a su punto isoeléctrico (isoelectroenfoque) utilizando tiras comerciales cubiertas por un gel de acrilamida. Para ello, las proteínas de la muestra deben ser totalmente solubilizadas, lo que se consigue generalmente con urea/tiourea, un detergente no iónico (CHAPS), un agente reductor (DTT) y una mezcla de anfolitos. Concluida la primera dimensión, la tira se carga sobre un gel desnaturalizante de poliacrilamida donde las proteínas se separan de acuerdo con su masa molecular. Así se obtiene un mapa bidimensional de manchas proteicas que podrá ser visualizado mediante distintas técnicas de tinción (azul de Coomassie, tinción con plata o Sypro Ruby). Los geles teñidos se escanean y digitalizan usando diferentes programas informáticos (ImageMaster, PD Quest) y se obtiene un «mapa» del conjunto de proteínas de una muestra en un gel. Para su identificación, las manchas de interés se recortarán del gel y las proteínas presentes serán digeridas por una enzima proteolítica altamente específica, usualmente tripsina, obteniéndose una mezcla de péptidos que será analizada por espectrometría de masas (MS). La comparación de sus espectros de masas frente a las librerías de espectros de péptidos trípticos existentes a través de motores de búsqueda como MASCOT o SEQUEST, permite identificar la proteína que dio lugar a dichos péptidos. Para ello, se realiza un análisis de MS simple (huella dactilar peptídica o PMF) que se ha de confirmar por un análisis de fragmentación peptídica o mediante espectrometría de masas en tándem (MS/MS)11,12 (fig. 3).
Proteómica cuantitativaProteómica cuantitativa con marcaje (2D-DIGE, i-TRAQ y SILAC) incluyendo separación de las proteínas (isoelectroenfoque y SDS-PAGE), visualización de las manchas proteicas y posterior identificación por espectrometría de masas.")
Uno de los aspectos claves en Investigación Biomédica es poder identificar y cuantificar las variaciones observadas entre individuos sanos y enfermos y a su vez los cambios observados durante los diferentes estadios de la patología con el fin de descubrir biomarcadores de pronóstico o diagnóstico. En este sentido, existen diferentes aproximaciones proteómicas cuantitativas: 1) basadas en electroforesis bidimensional, destacando la metodología 2-dimensional difference gel electrophoresis (2D-DIGE), y 2) aproximaciones basadas en cromatografía líquida (LC) acoplada a espectrometría de masas (MS) empleando marcas tanto a nivel proteico como peptídico; es el caso de las metodologías isobaric tags for relative and absolute quantitation (iTRAQ) y stable isotope-labeling with amino acids in cell culture (SILAC).
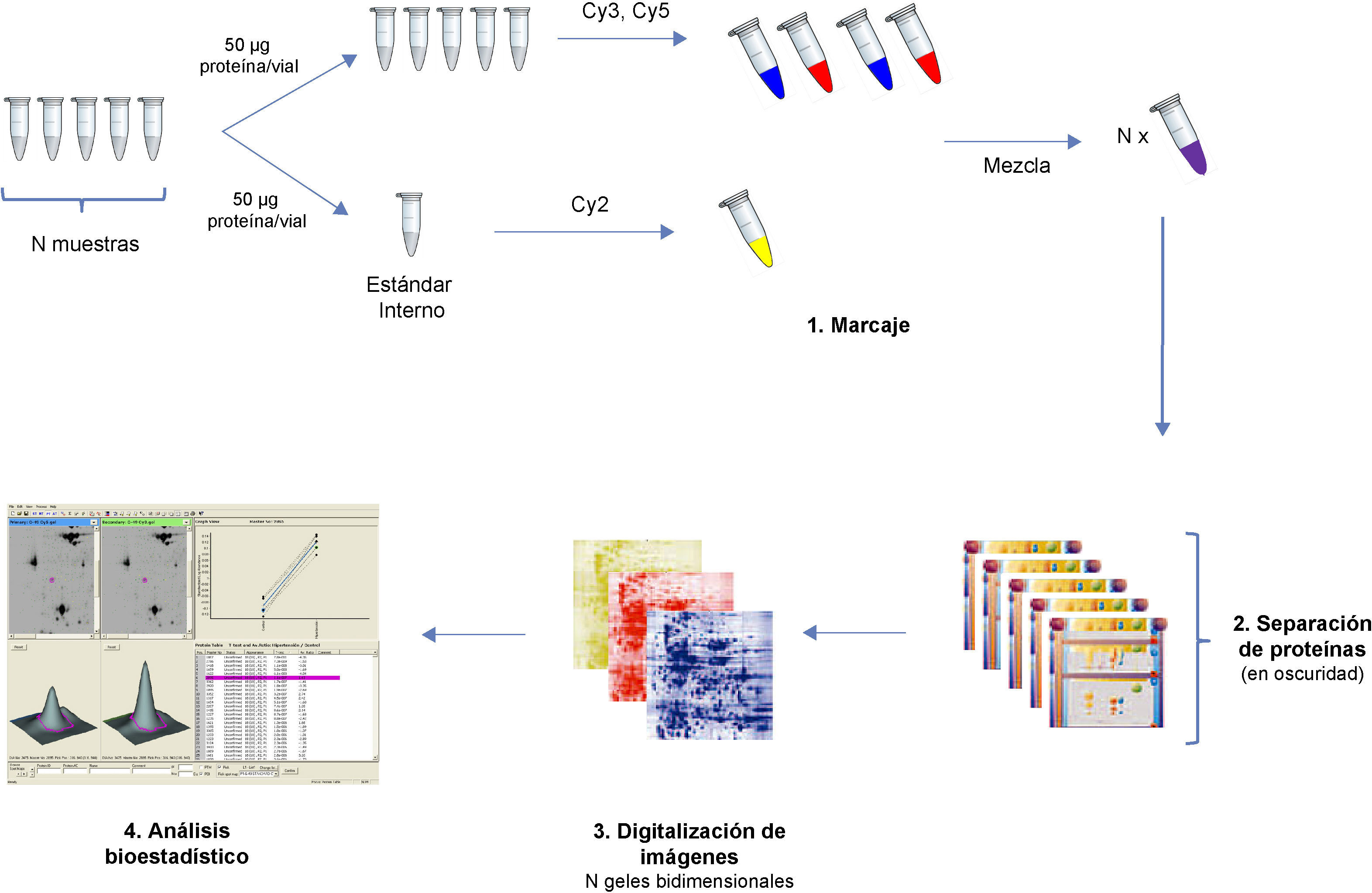
Electroforesis bidimensional diferencial 2D-DIGELa tinción de geles 2D con plata o azul Coomassie para hacer estudios comparativos presenta la desventaja de tener que realizar un gel por muestra, lo que se traduce en un gran número de geles cuya reproducibilidad es difícil de conseguir para obtener diferencias estadísticamente significativas. Además, el rango dinámico lineal de tales aproximaciones o la sensibilidad no siempre se ajustan a los requerimientos de nuestras muestras. Esto queda resuelto con el desarrollo de la electroforesis bidimensional diferencial en gel (2D-DIGE) una técnica basada en el marcaje de las proteínas de cada muestra previo a su separación con una de las tres sondas fluorescentes disponibles (Cy2, Cy3 y Cy5)13.
En cada gel se analizan simultáneamente hasta dos muestras marcadas con dos de los fluorocromos y una alícuota del «pool» marcada con el fluorocromo restante, que debe de estar presente en todos los geles. Este «pool» está constituido por una mezcla de todas las muestras que componen el estudio, lo cual permite extrapolar los resultados comparativos de gel a gel, reduciendo las diferencias debidas a las variaciones experimentales extrínsecas a la patología, asegurando la superposición de las manchas proteicas y proporcionando un incremento de la reproducibilidad y la fiabilidad del análisis de abundancia diferencial entre muestras. El procedimiento de análisis corresponde al de una electroforesis bidimensional convencional14. Las imágenes de los geles son digitalizadas empleando un escáner de fluorescencia, obteniendo las imágenes correspondientes a cada fluorocromo y analizadas estadísticamente mediante un software informático (DeCyder)15 que detecta automáticamente cada una de las manchas correspondientes a las proteínas resueltas y realiza un solapamiento de las mismas entre todas las imágenes del experimento. Las variaciones significativas en los perfiles de abundancia proteica se pueden identificar mediante análisis bioestadísticos y dichos perfiles se pueden agrupar en función de sus similitudes mediante un análisis de componentes principales (PCA) o un análisis de alta abundancia (HCA) entre otros. Un resumen del procedimiento 2D-DIGE se esquematiza en la figura 4.
Cromatografía líquida multidimensional i-TRAQ®: marcaje de las muestras con fluorocromos, separación proteica en 2-DE, visualización de imágenes y análisis estadístico.")
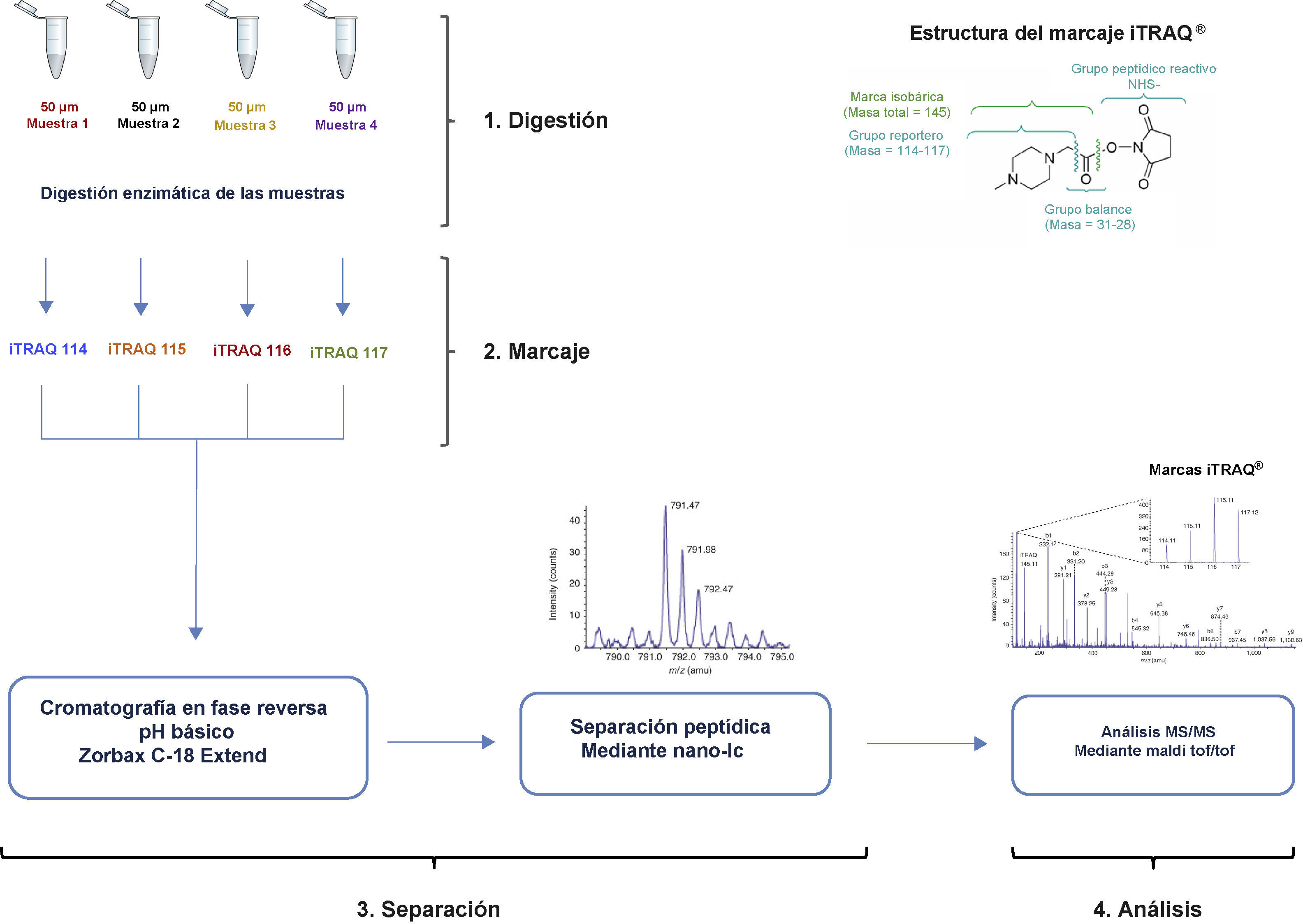
El marcaje iTRAQ® (isobaric Tags for Relative and Absolute Quantification, Applied Biosystems) es una metodología que permite cuantificar y observar modificaciones proteicas mediante un marcaje químico. A diferencia del 2D-DIGE, no se basa en geles sino en una digestión peptídica de las muestras proteicas, posterior marcaje de los péptidos resultantes y análisis final por LC-MS/MS. Emplea un marcaje con moléculas isobáricas (de igual masa nominal) que constan de tres regiones: 1) una activa de unión a péptido a través de un grupo N-hidroxi-succimida, capaz de reaccionar con el grupo amino N-terminal de los péptidos y el grupo amino de la cadena lateral de las lisinas; 2) una región reportera de masa variable, que identifica cada una de las muestras a analizar sin alterar la carga ni la eficiencia de ionización del péptido marcado (los cuatro grupos reporteros aparecen en el espectro de masas en m/z 114, 115, 116 y 117), y 3) un grupo balance para que la masa total sea igual a 145Da en todos los marcajes16 (fig. 5).

Esquema de flujo de trabajo para llevar a cabo un estudio proteómico cuantitativo mediante cromatografía líquida multidimensional i-TRAQ®. La mezcla proteica se digiere y los péptidos resultantes, una vez marcados, se separan por cromatografía líquida y se analizan por espectrometría de masas.
En un experimento iTRAQ®, las muestras proteicas son digeridas con peptidasas y los péptidos resultantes de cada muestra se marcan con los diferentes reactivos iTRAQ® por separado (pueden llegar a marcarse de 4 a 8 muestras por experimento). Una vez marcadas, las muestras se combinan y analizan por LC-MS/MS. Como el marcaje es isobárico, los péptidos idénticos pero con una marca diferente según la muestra de la que procedan, no se distinguen cuando son analizados por MS. En cambio, mediante espectrometría de masas en tándem (MS/MS) se consigue, por un lado, la identificación de las proteínas presentes mediante la búsqueda en bases de datos y por otro lado, el análisis semi-cuantitativo de las mismas por comparación de las intensidades relativas de los iones reportero, que son detectables en la primera parte del espectro de fragmentación de cada péptido. Variaciones en dichas intensidades se traducen en una variación en los niveles de la proteína que dio lugar a dicho péptido.
Marcaje de aminoácidos con isotopos estables en medio celular (SILAC)SILAC es una técnica de marcaje de proteínas in-vivo para su posterior identificación y cuantificación por MS. Este método se basa en la incorporación de aminoácidos marcados con isótopos estables (13C, 15N). Así, en un mismo experimento, dos poblaciones celulares se cultivan en medios de cultivos idénticos con la salvedad de que uno va marcado con la forma ligera y otro con la forma pesada de un aminoácido en particular (por ejemplo, L-lisina marcada con 12C y L-lisina marcada con 13C). De esta manera las células en lugar de sintetizar los aminoácidos naturales, sintetizarán los aminoácidos marcados, es decir, después de una serie de divisiones celulares, cada aminoácido natural, será reemplazado por su isótopo marcado analógico. Dado que no hay casi ninguna diferencia química entre el aminoácido marcado y los isótopos naturales las células se comportan exactamente igual que la población de células cultivadas con los aminoácidos normales.
Proteómica cuantitativa sin marcajeSe trata de una metodología muy utilizada últimamente debido a que no requiere ningún marcaje. Permite realizar la cuantificación de proteínas en base a todos los péptidos y no sólo a aquellos correctamente marcados. Aunque se trata de un método semi-cuantitativo, evita el elevado coste de los reactivos de marcaje y/o la síntesis de péptidos estándar marcados isotópicamente. Uno de los primeros métodos empleados es el de contaje de péptidos (spectral counting), que se basa en la existencia de una correlación entre la frecuencia con la que el espectrómetro de masas en tándem fragmenta un ión y la proteína de la que procede dicho ión. Es decir, al aumentar la abundancia de una proteína en una muestra dada aumenta el número de péptidos únicos identificados. Posteriormente, se han desarrollado métodos de cuantificación basados en el área de los picos de los iones en el espectro de masas, de tal forma que el área resultante resulta equivalente a su abundancia en la muestra. Existen varios programas informáticos que permiten estandarizar y normalizar los resultados obtenidos por esta metodología: DeCyder MS (GE Healthcare), SIEVE (Thermo electron), Elucidator (Rosetta), ProteinLyx (Waters), Scaffold (Thermo).
Una plataforma de análisis proteómico de muestras biológicas orientada al descubrimiento de potenciales biomarcadores es el sistema Protein Chip/Surface Enhanced Laser Desorption/Ionization-Time of Flight (SELDI-TOF) que combina la retención específica de proteínas sobre biochips con la identificación de las proteínas mediante MS. Las proteínas son capturadas sobre la superficie sólida de un chip en el que se han depositado soportes de distinta naturaleza química (aniónica, catiónica, hidrofóbica, hidrofilíca, iones metálicos, etc.) o bioquímica (anticuerpos, receptores, DNA, enzimas, etc.). Las proteínas quedan retenidas en las distintas zonas en función de sus características y las del soporte, mediante adsorción, partición, afinidad, etc. Son, por tanto, chips cromatográficos capaces de retener grupos particulares de proteínas a partir de mezclas complejas de las mismas, como suero/plasma, orina, o lisados celulares. Las proteínas en el chip son ionizadas y vaporizadas mediante el láser y sus masas moleculares (m/z) determinadas en función del tiempo de vuelo. El resultado del análisis representa la abundancia (intensidad) frente a la masa molecular de las proteínas detectadas, a modo de perfiles proteicos facilitando la comparación entre múltiples muestras distintas. El análisis multivariante, incluido en el software del equipo, permite identificar patrones o perfiles proteicos específicos asociados con la enfermedad a nivel diagnóstico, pronóstico o de recuperación, con capacidad discriminante respecto a poblaciones de individuos sanos u otras patologías17–19.
Existe otra metodología de cuantificación libre de marcaje que se basa en el empleo de arrays de proteínas. Dentro de estos se encuentran por un lado, los arrays de anticuerpos con un interés específico en una patología determinada cuyas proteínas de interés pueden ser cuantitativamente analizadas y de modo comparativo. Por otro lado, los arrays de fase reversa que permiten determinar las proteínas implicadas en rutas de señalización pudiendo determinar el estado de fosforilación de las proteínas implicadas en dichas rutas intracelulares20. La utilización de los arrrays de fase reversa permite obtener un tipo de información molecular único e individualizado de cada muestra, y por tanto de cada paciente. Se obtienen de esta manera, perfiles de señalización intracelular, incluyendo especialmente las modificaciones postraduccionales (fosforilaciones), datos que no son accesibles mediante técnicas genómicas.
Monitorización por reacción seleccionada (SRM)La validación de los potenciales biomarcadores descubiertos en la fase de «screening» es una de las principales limitaciones para su aplicación directa en clínica. Estrategias y metodologías diferentes a las empleadas en esa primera fase son necesarias para garantizar la fiabilidad de los resultados. En este sentido, especial atención merece la metodología de cuantificación libre de marcaje «selected reaction monitoring»(SRM) por su elevada sensibilidad e idoneidad como estrategia de validación de potenciales biomarcadores. Requiere el empleo de un espectrómetro de masas de triple cuadrupolo (QQQ). El primer paso para cuantificar una proteína de interés en varias muestras sería la selección de péptidos proteotípicos, seguido de la selección de transiciones a medir según los fragmentos característicos de dichos péptidos. Por péptido proteotípico entendemos aquel que inequívocamente identifica la proteína, se observa con alta frecuencia en un experimento de MS, genera preferentemente varios fragmentos de intensidad destacada (frente a muchos fragmentos de intensidad similar), no presentan modificaciones pos-traslacionales y, en principio, no es susceptible de modificación química. De cara a escoger las transiciones más adecuadas para la cuantificación de la proteína, tendremos en cuenta los fragmentos más intensos en base a experimentos previos. Mediante herramientas bioinformáticas se pueden calcular teóricamente los péptidos proteotípicos y las transiciones posibles que origina una proteína. Sin embargo, habrá que hacer una selección dependiendo del número de proteínas y péptidos correspondientes a cuantificar por experimento, ya que un número demasiado elevado conllevaría una mala definición del pico cromatográfico con la consecuente pérdida de información, ya que a mayor tiempo empleado en la medida de transiciones menor es el número de puntos que permiten definir el pico. Para evitar una medida errónea por medida de señales inespecíficas que coinciden en masa con las transiciones de interés, será necesario adquirir varias transiciones por péptido y que éstas solapen en el tiempo (el péptido que las origina eluye a un tiempo determinado que será al que aparezcan las transiciones). Mediante MS/MS del precursor, se confirma la identificación de la proteína.
Desde un punto de vista instrumental, en un experimento SRM típico Q1 transmite a Q2 una sola masa (m1), correspondiente al péptido tríptico completo. Q2 actúa de célula de colisión (CID) y se optimiza para que al fragmentar la molécula se produzca algún fragmento diagnóstico del mismo. Q3 opera de forma que sólo transmite dicho fragmento (m2 o m3). Así, seleccionando las transiciones más sensibles (m1 a m2 y m1 a m3), se obtiene una señal para cada una de ellas y en función de su intensidad relativa se podrán cuantificar cambios a nivel de abundancia proteica21. Para realizar una cuantificación por SRM debemos conocer la/s proteína/s de interés, lo que no permite su empleo para análisis diferencial en la fase de descubrimiento de biomarcadores. En la actualidad esta técnica se está imponiendo como técnica de validación de biomarcadores debido a su gran selectividad22, así como por el manejo del gran número de muestras que permite.
InmunoproteómicaDe una manera más específica podemos hablar de la immunoproteómica para describir el estudio de grandes conjuntos de proteínas implicados en la respuesta inmune (inmunoma)23, siendo sus principales aplicaciones: 1) el aislamiento e identificación mediante MS de proteínas pertenecientes al complejo principal de histocompatibilidad (MHC); 2) purificación e identificación de proteínas antigénicas de unión a anticuerpos específicos u otros reactivos de afinidad, y 3) identificación de las proteínas y las vías moduladas por un microorganismo específico, enfermedad infecciosa o toxina. Dentro del análisis del conjunto de proteínas incluidas en el MHC, una de las aplicaciones más interesantes de la inmunoproteómica consiste en el estudio de la respuesta serológica frente a un agente infeccioso y de las bases moleculares de su patogenicidad. Al igual que ocurre con otras aplicaciones proteómicas, la inmunoproteómica constituye una excelente herramienta para el descubrimiento de potenciales biomarcadores para las enfermedades infecciosas especialmente mediante un análisis proteómico del suero (SERPA, serological proteome analysis) en el cual se utiliza una electroforesis bidimensional clásica (2-DE) para la separación de las proteínas que son posteriormente transferidas a una membrana sobre la que aplican sueros de pacientes infectados y de controles pudiéndose detectar proteínas inmunogénicas mediante western-blot y que serán finalmente identificadas empleando técnicas de espectrometría de masas comentadas anteriormente24,25. A través de estos estudios la inmunoproteómica, como aplicación específica de la proteómica, ha proporcionado un elevado número de potenciales biomarcadores de diagnóstico, pronóstico, predicción y/o monitorización de diferentes enfermedades infecciosas, así como posibles dianas terapéuticas para el diseño de futuras inmunoterapias o vacunas frente a estas26,27.
Proteómica de fluidos biológicosEstudio proteómico del plasmaEl plasma sanguíneo es, junto con la orina, la muestra biológica más accesible y que más ampliamente ha sido utilizada históricamente en la práctica clínica28. Cuando se lleva a cabo un estudio proteómico de plasma, el objetivo principal es la búsqueda de aquellas proteínas que sirvan como biomarcadores de diagnóstico y pronóstico de una enfermedad determinada asociada a procesos fisiológicos y patológicos concretos29. Desde este punto de vista, las proteínas que tienen un mayor interés son aquellas que se encuentran en muy baja concentración30. Sin embargo, el amplio rango de concentraciones de las proteínas del plasma (mg/ml a pg/ml) hace que no sea una tarea fácil y se requiere cuando menos una depleción total o parcial de las mayoritarias.
La proteómica ofrece, por tanto, un gran potencial para el descubrimiento de nuevos biomarcadores que sean trasladables a la práctica clínica. Hay que tener en cuenta que actualmente en los laboratorios clínicos se realiza rutinariamente la determinación del contenido global de proteínas séricas (proteínas totales), sin embargo, la información que esta prueba nos proporciona es limitada puesto que únicamente refleja cambios en las proteínas más abundantes con alteraciones significativas en casos de hipergammaglobulinemias, estados hipo/hipervolémicos, deficiencias nutritivas, hepatopatías, procesos inflamatorios y trastornos renales, entre otros. También se lleva a cabo la cuantificación de proteínas plasmáticas específicas (albúmina, PCR, β2-microglobulina, etc.) y la medida de la actividad de enzimas plasmáticas (CK, CKMB, AST, ALT, GGT, etc.). Ambas se emplean desde hace años para el diagnóstico y seguimiento de un gran número de enfermedades (hepáticas, pancreáticas, cardiacas, etc.) sin embargo, al igual que ocurre con la determinación de proteínas totales, son pruebas poco específicas de un estado patológico concreto.
Por tanto, la determinación de proteínas minoritarias mediante estudios proteómicos supondría un gran avance para el diagnóstico precoz de muchas enfermedades con respecto a la situación que existe en la actualidad.
Estudio proteómico en orinaEl uso de biomarcadores en orina para el diagnóstico de enfermedades es una práctica habitual en clínica debido a que: 1) es una muestra fácil de obtener de manera no invasiva y cuenta con una gran disponibilidad; 2) la composición proteica y peptídica de la orina es menos compleja que la de otras muestras y cuenta con una mayor estabilidad, y 3) la composición del proteoma de la orina puede reflejar directamente cambios en funciones del riñón y del tracto urogenital31. Sin embargo, la orina también presenta ciertas dificultades a la hora de emplearla como fuente en la búsqueda de biomarcadores ya que su composición puede variar de manera significativa entre distintos individuos y entre los distintos días/horas de colección de la muestra, incluso de un mismo individuo lo cual requiere en la práctica clínica el uso de intervalos de referencia muy amplios en comparación con las determinaciones séricas.
Al igual que en el caso del plasma y análisis de fluidos biológicos en general, hay dos formas de abordar una búsqueda de biomarcadores en orina. La primera implicaría la pre-selección de posibles candidatos (proteínas tubulares, citocinas, factores de crecimiento, mediadores inflamatorios, etc.) apoyándonos en trabajos previos que indiquen una posible conexión con la patología en estudio como el uso de la β-gonadotropina coriónica en la orina de mujeres como base de los test de embarazo comerciales. La segunda abordaría un análisis proteómico que consiste en hacer un barrido de todas las proteínas de la orina sin sesgo en busca de aquellas que pudieran permitirnos establecer una conexión con la enfermedad. La organización HUPO cuenta con una iniciativa particular centrada en el proteoma del riñón y la orina, poniendo nuevamente de manifiesto el potencial de este fluido como fuente de biomarcadores. Novedosos estudios de proteómica en orina están proporcionando información sobre proteínas que podrían ser de gran valor para el diagnóstico y monitorización de enfermedades sistémicas, urogenitales y renales32.
Se considera que las proteínas de orina se pueden clasificar en tres fracciones: proteínas solubles, proteínas sedimentadas y exosomas, que pueden ser aisladas mediante centrifugación secuencial. Cada una de estas fracciones contiene poblaciones proteicas distintas. Aunque el estudio de exosomas urinarios ha aumentado en los últimos años, la mayoría de los estudios llevados a cabo en la búsqueda de biomarcadores en orina se han centrado en la fracción de proteínas solubles y se ha prestado especial atención a enfermedades renales y del tracto urogenital. Existen estudios en daño renal agudo, enfermedad glomerular, carcinoma renal, de vejiga y de próstata33,34. Cambios en el proteoma urinario pueden estar relacionadas con enfermedades sistémicas asociadas a pequeñas proteínas circulantes y péptidos marcadores capaces de atravesar el filtro glomerular. Existen publicaciones en las que se han identificado biomarcadores urinarios predictivos para pancreatitis aguda, apnea del sueño, cáncer de ovario temprano y cáncer de pulmón de célula no pequeña35,36. Por tanto, en el futuro la orina podría ser empleada para hacer barridos proteicos que permitan detectar desordenes sistémicos que no tienen por qué estar limitados a afecciones renales.
Estudio proteómico de otras muestras biológicas de relevancia clínicaUna de las principales ventajas de las técnicas proteómicas radica en que pueden aplicarse al estudio de cualquier muestra (líquido biológico, células, tejidos, etc.) cuyo proteoma se desee conocer. Así, aunque la búsqueda de nuevos biomarcadores diagnóstico y pronóstico de las enfermedades humanas se ha llevado a cabo mayoritariamente en suero/plasma y orina por ser estas las muestras biológicas más accesibles y más comúnmente utilizadas en la práctica clínica, también se han llevado a cabo estudios proteómicos en otras muestras biológicas diferentes como líquido cefalorraquídeo, líquido amniótico y diversos tejidos humanos para el estudio de enfermedades concretas.
Dada la complejidad proteica del plasma, es de gran interés llevar a cabo estudios complementarios de proteínas a nivel de secretoma de explantes tisulares como sub-proteoma compuesto por las proteínas liberadas directamente por el tejido de interés y que serán analizadas directamente en esta fracción, evitando así la dilución que supondría su análisis directo en el plasma. Para confirmar el origen de las proteínas como procedentes del tejido analizado y no de una fuente externa de contaminación por parte del plasma, son útiles las estrategias de marcaje con aminoácidos marcados en el medio de cultivo. De este modo, en el análisis del secretoma por espectrometría de masas todas aquellas proteínas sintetizadas por el tejido y posteriormente secretadas incorporarán los aminoácidos marcados en sus secuencias y podrá así ser validado su origen37.
El estudio del líquido cefalorraquídeo (LCR) mediante técnicas proteómicas se ha orientado a la búsqueda de biomarcadores en enfermedades neurodegenerativas como el Alzheimer, el Parkinson y la esclerosis múltiple38. En todos los casos se han encontrado paneles de proteínas diferencialmente abundantes entre los grupos controles y los pacientes. El objetivo del estudio del líquido amniótico es buscar biomarcadores en aquellas enfermedades que afectan al feto y que requieren un diagnóstico precoz. Así se han llevado a cabo estudios para la búsqueda de perfiles proteicos característicos que permitan detectar la ruptura prematura de membrana antes del comienzo de la infección ya que es la causante a posteriori de muchos partos prematuros39. También se llevan a cabo estudios para la determinación de nuevos biomarcadores en cromosomopatías frecuentes como el síndrome de Down donde se han descrito diferencias cuantitativas en la abundancia de diversas proteínas en líquido amniótico40. El descubrimiento de nuevos biomarcadores en suero/plasma materno constituiría un gran avance en estas enfermedades y permitiría sustituir las técnicas invasivas existentes actualmente como la amniocentesis, por determinaciones en suero/plasma materno que complementarían el triple screening (alfafetoproteína, gonadotropina, estriol) utilizado actualmente en la práctica clínica para el cribado de síndrome de Down. Otro tipo de muestra biológica más recientemente empleado para estudios proteómicos es la saliva. Este fluido constituye un ultrafiltrado del plasma de fácil obtención y por lo tanto resulta una opción muy interesante en la búsqueda de biomarcadores41–43.
La organización HUPO además del proyecto del proteoma del plasma y del proteoma del riñón y la orina, cuenta con varias iniciativas particulares centradas en distintas patologías, tales como el Human Brain Proteome Project (HBPP, ‘proyecto de proteoma cerebral’), el Human Liver Proteome Project (HLPP, ‘proyecto de proteoma hepático’) y la CardioVascular Initiative (CVI, ‘iniciativa cardiovascular’), entre otros. La importancia de estos proyectos se fundamenta en la necesidad de conocer de una manera detallada el proteoma de los órganos o fluidos biológicos cuyo estudio contribuya directamente a ampliar el conocimiento existente sobre los mecanismos base de la enfermedad así como al descubrimiento de nuevas dianas de interés a todos los niveles (diagnóstico, estudio de la enfermedad, susceptibilidad, prevención, selección de terapias, seguimiento de los tratamientos, etc.).
Por último, es importante destacar la relevancia clínica que se esconde detrás de todo avance tecnológico. A modo de ejemplo, el desarrollo de la técnica microdisección por láser ha permitido aislar regiones específicas de tejido (por ejemplo las distintas capas arteriales, íntima y media, implicadas directamente en la formación de placa de ateroma) y mediante su combinación con la metodología 2D-DIGE, anteriormente descrita, ha hecho posible el conocimiento de proteínas directamente implicadas en la formación y desarrollo de la aterosclerosis44. Indudablemente estos avances suponen una paso adelante para la comprensión de la fisiopatología de las enfermedades que permita descubrir nuevos biomarcadores de enfermedad.
ConclusionesLa práctica clínica actual requiere nuevos biomarcadores sensibles y específicos que mejoren el diagnóstico temprano de las enfermedades. Es necesario establecer nuevas dianas terapéuticas que mejoren o hagan posible, en algunos casos, la curación de determinadas enfermedades y se requiere además, el descubrimiento de nuevos biomarcadores que permitan monitorizar de manera individualizada la respuesta terapéutica a una determinado tratamiento. La proteómica basada en la identificación de proteínas mediante espectrometría de masas ofrece un enorme potencial en cada uno de estos puntos. El descubrimiento de nuevos biomarcadores pronóstico y diagnóstico es el objetivo fundamental de gran parte de las investigaciones proteómicas actuales. Además, su descubrimiento podría derivar en la implementación de nuevos métodos diagnósticos en la práctica clínica así como en el descubrimiento de nuevas dianas terapéuticas más efectivas que las actuales.
AutoríaGloria Álvarez-Llamas y María G. Barderas contribuyen de igual manera en este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
